
FDA Issues Draft Guidance on Continuous Manufacturing

The FDA has issued draft guidance for continuous manufacturing of drug substances and drug products. Industry feedback is due by mid-December. What does the FDA say?
The FDA and continuous manufacturing
Earlier this month (October 2021), the US Food and Drug Administration issued draft guidance, Q13 Continuous Manufacturing of Drug Substances and Drug Products. The draft guidance, which was prepared under the support of the International Council for Harmonization (ICH), describes scientific and regulatory considerations for the development, implementation, operation, and lifecycle management of continuous manufacturing. The ICH is a non-binding body that seeks to harmonize international standards for the bio/pharmaceutical industry, including on issues related to manufacturing and quality.
ICH Q13 was endorsed by the ICH Assembly in June 2021 and is now under consideration by the regulatory authorities, including the FDA, which participate in the ICH. The FDA’s draft guidance applies to continuous manufacturing of drug substances and drug products for chemical entities and therapeutic proteins. The draft guidance is applicable to new products (e.g., new drugs, generic drugs, biosimilars) and the conversion of existing products using batch manufacturing to continuous manufacturing.
Building on existing ICH Quality guidelines, the draft guidance provides clarification on continuous manufacturing concepts and describes scientific approaches and regulatory considerations specific to continuous manufacturing of drug substances and drug products. The FDA is seeking feedback from stakeholders, including the bio/pharma industry, on the draft guidance by December 13, 2021.
Regulatory support key for the adoption of continuous manufacturing
The FDA has long supported continuous manufacturing and has taken steps to facilitate the bio/pharmaceutical industry’s implementation of continuous manufacturing. The FDA first put forth its support for continuous manufacturing in 2002 with the agency’s initiative, Pharmaceutical Current Good Manufacturing Practices for the 21st Century, as a means to modernize pharmaceutical manufacturing and enhance product quality. This initiative included putting forth a science- and risk-based approach to pharmaceutical manufacturing and regulatory oversight through a Quality-by-Design approach as well as by encouraging new technologies, such as process analytical technology (PAT) and continuous manufacturing.
Support by the FDA and other regulatory agencies for continuous manufacturing is crucial. With the industry’s installed manufacturing base still in batch manufacturing and further investment required in equipment, analytical technology, such as PAT, and technical training for continuous manufacturing, the industry has been proceeding on a measured basis in adopting continuous manufacturing.
To date, the FDA has approved 10 drugs that use a continuous manufacturing process. In 2020, the FDA approved four drugs using continuous manufacturing, which included the first regulatory application using continuous manufacturing for an active pharmaceutical ingredient, the first continuous biomanufacturing process, as well as two different marketed products using semi-continuous manufacturing processes. The first approval for a drug made using a continuous manufacturing process was in 2015 for Vertex’s Orkambi (lumacaftor/ivacaftor), a drug for treating cystic fibrosis. Since then, Vertex has had two other drugs approved using continuous manufacturing, also to treat cystic fibrosis: Symdeco/Symkevi (tezacaftor/ivacaftor and ivacaftor) and Trikafta (elexacaftor/tezacaftor/ivacaftor and ivacaftor). Johnson & Johnson received approval for its HIV drug, Prezista (darunavir), which was the first drug that the FDA allowed to be switched from batch manufacturing to continuous manufacturing. Other FDA-approved drugs using continuous manufacturing include Pfizer’s Daurismo (glasdegib) for treating acute myeloid leukemia and Lilly’s Verzenio (abemaciclib) for treating metastatic breast cancer.
Inside the FDA’s draft guidance
The FDA’s draft guidance is aligned with the ICH guideline, which provides global harmonization for continuous manufacturing regulatory approaches and encourages broader adoption of this technology.
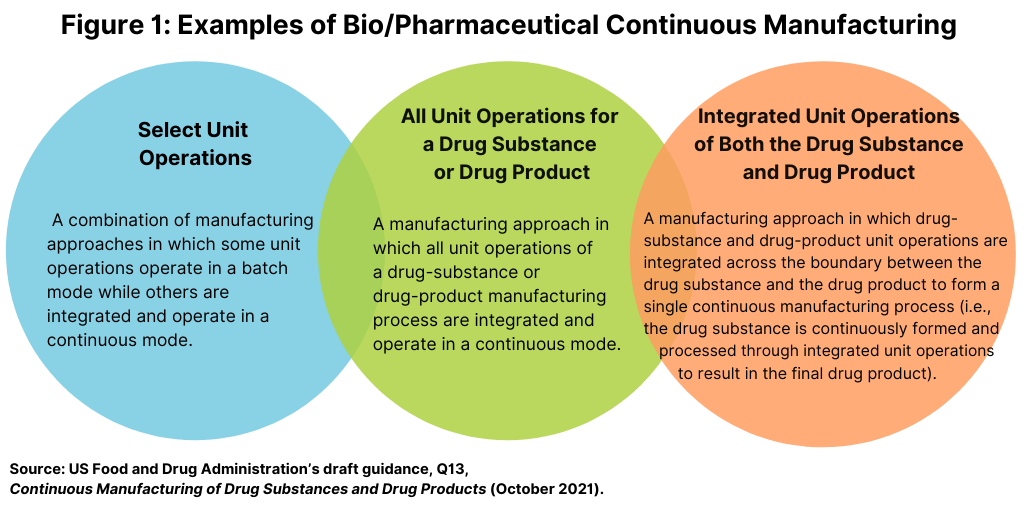
Continuous manufacturing involves the continuous feeding of input materials into, the transformation of in-process materials within, and the concomitant removal of output materials from a manufacturing process. While this description may apply to an individual unit operation (e.g., tableting, perfusion bioreactors), the FDA draft guidance focuses on the integrated aspects of a continuous manufacturing system in which two or more unit operations are directly connected (see Figure 1). In this context, any changes made in a unit operation of continuous manufacturing may have a direct and often immediate impact on downstream and upstream unit operations.
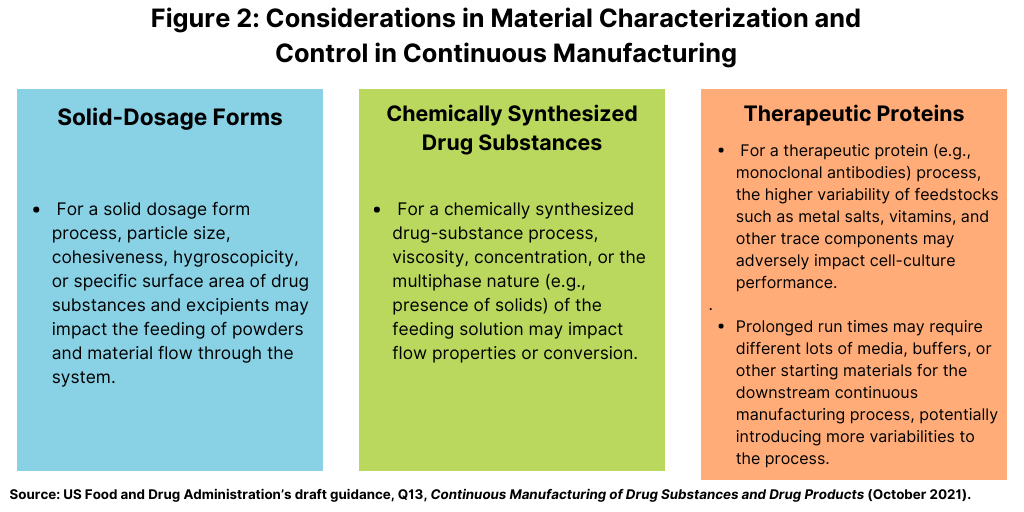
As outlined in the FDA’s draft guidance, material attributes can impact various aspects of continuous operation and performance, such as material feeding, process dynamics, and output-material quality. Understanding the impact of material attributes and their variability on process performance and product quality is important for the development of the control strategy. Input materials may require evaluation and control of attributes beyond those typically considered for a material specification used in batch manufacturing (see Figure 2).
The control strategy of a continuous manufacturing process is designed to ensure that output materials made over the run time are of the desired quality as highlighted in the FDA’s draft guidance. The control strategy should describe the relevant controls and approaches used during the manufacturing and the operational aspects of the continuous manufacturing process (see Figure 3).

Why continuous manufacturing?
Although batch manufacturing prevails in the bio/pharmaceutical industry, continuous manufacturing holds the potential for improved process control and reduced and more flexible manufacturing footprints.
In issuing its draft guidance, the FDA highlighted several key benefits of continuous manufacturing as outlined below:
- Eliminates manual handling and human error;
- Increases quality assurance through online monitoring and control;
- Reduces manufacturing time and increases efficiency;
- Reduces capital costs by using smaller equipment and less manufacturing space;
- Responds more nimbly in the event of a drug shortage; and
- Allows manufacturers to tailor drug production to fit the needs of precision medicines.
Other measures in the US
The FDA and other policy makers are seeking to advance continuous manufacturing. Earlier this month (October 2021), the US House of Representatives passed a bill, The National Centers of Excellence in Advanced and Continuous Pharmaceutical Manufacturing Act of 2021 (H.R.4369), which directs the FDA to designate qualified institutions of higher education as National Centers of Excellence in Continuous Pharmaceutical Manufacturing and to provide grants to the centers. Each designated center must conduct research on continuous manufacturing technologies and must share information from such research with the FDA. The bill was introduced in the US Senate and was referred to the Senate Committee on Health, Education, Labor, and Pensions for further consideration.
In addition to issuing guidance, under its Emerging Technology Program, the FDA seeks to interact with companies to facilitate the adoption of new technologies, including continuous manufacturing. The program recognizes that bio/pharmaceutical companies may have concerns that using such technologies could result in delays while FDA reviewers familiarize themselves with the new technologies and determine how they may be evaluated within the existing regulatory framework. To address these concerns, the FDA’s Office of Pharmaceutical Quality created the Emerging Technology Program to promote the adoption of innovative approaches to pharmaceutical product design and manufacturing. Through the program, industry representatives can meet with Emerging Technology Team members to discuss, identify, and resolve potential technical and regulatory issues regarding the development and implementation of a novel technology prior to filing a regulatory submission.




