FDA & Remote Inspections: What Is Next?

The FDA has issued a draft guidance to lay out its current thinking on remote regulatory assessments, an approach it took when onsite inspections were not possible due to the COVID-19 pandemic. What’s in the plan?
FDA’s draft guidance on remote regulatory assessments.
Late last month (July 2022), the US Food and Drug Administration (FDA) released a draft guidance on the expanded use of remote regulatory assessments (RRAs) and how the FDA generally intends that this tool, once finalized, will be incorporated across all FDA-regulated products beyond the current COVID-19 public health emergency. The FDA is seeking feedback on the draft guidance by September 23, 2022.
Over the last two years, the FDA has performed more than 1,470 domestic and more than 600 foreign entity establishment RRAs. These remote assessments of FDA-regulated establishments and/or their records are used by the FDA to help determine compliance with applicable FDA requirements, inform regulatory decisions, and verify information submitted to the agency.
The draft guidance, Conducting Remote Regulatory Assessments Questions and Answers, is intended to provide transparency to stakeholders about how RRAs may be used and to promote consistency in the way RRAs are conducted. The draft guidance covers voluntary and statutorily authorized RRAs but does not change the core requirements of inspections and pre- and post-market authorities. Except for RRAs for establishments required to comply with the Foreign Supplier Verification Programs regulation, an RRA does not function as an inspection.
“We intend to continue to use RRAs, as appropriate, according to a risk-based approach that best protects public health,” said Robert M. Califf, FDA’s Commissioner of Food and Drugs, in a July 22, 2022, statement. “For example, when we are unable to deploy in-person staff due to travel restrictions, we may determine that an RRA is an appropriate tool. RRAs may also be used to efficiently assess establishments identified in product applications when these establishments have a prior history of compliance. Where appropriate, RRAs allow the FDA to review information such as livestreams and records provided by a company without going onsite, which can make better use of limited agency resources and give industry more scheduling flexibility.”
Figure 1 describes key highlights of the draft guidance, including why and how the FDA may initiate or request an RAA and the types of information that may be requested by the FDA.
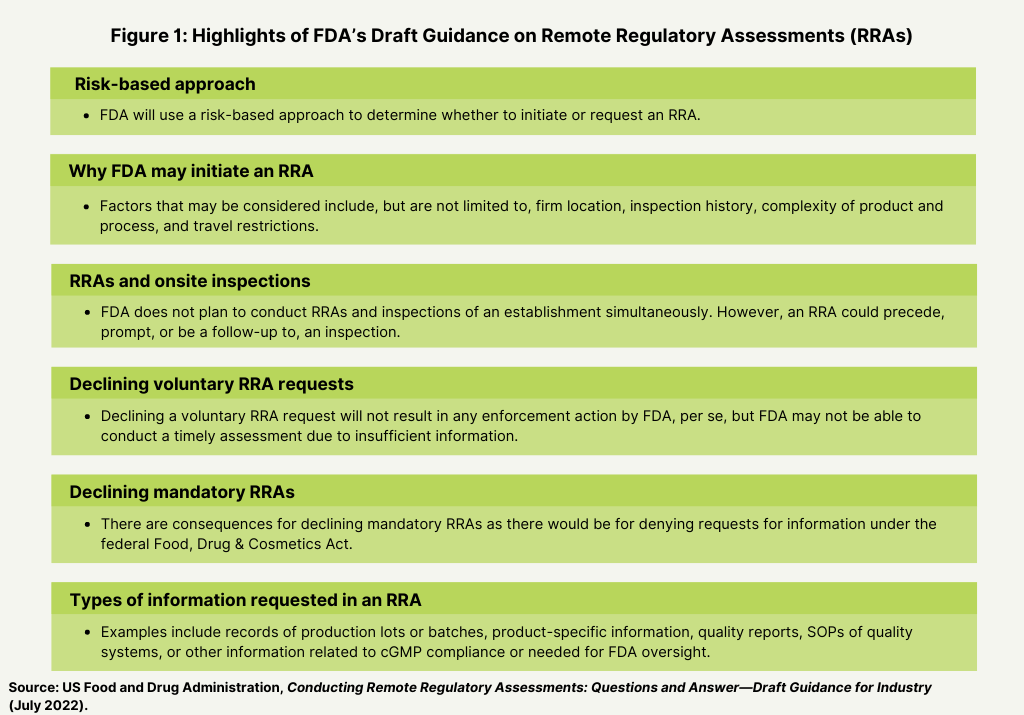
The FDA says it will use a risk-based approach to determine whether to initiate or request an RRA. Factors that may be considered include, but are not limited to, firm location, inspection history, complexity of product and process, and travel restrictions. The FDA says it does not plan to conduct RRAs and inspections of an establishment simultaneously. However, an RRA could precede, prompt, or be a follow-up to, an inspection. An RRA is conducted remotely by FDA staff without FDA staff present at an establishment conducting an inspection. When an RRA precedes an inspection, the FDA says it will generally conclude the RRA prior to initiating the inspection. The FDA may combine any information gained from the RRA with any resulting observations from the subsequent inspection. In such circumstance, FDA would confirm any observations from the RRA during the inspection before including them on the Form FDA 483 Inspectional Observations. Additionally, FDA may conduct an RRA following an inspection in order to conduct follow-up activities with the establishment or to assist in verifying corrective actions, if appropriate.
In addition, the FDA says that declining a voluntary RRA request will not result in any enforcement action by FDA, per se, but that the agency may not be able to conduct a timely assessment due to insufficient information. There are consequences, however, for declining mandatory RRAs as there would be for denying requests for information under the federal Food, Drug & Cosmetics Act.
Examples of the type of information that may requested in an RAA include records of production lots or batches, product-specific information, quality reports, standard operating procedures of quality systems, or other information related to cGMP compliance or needed for FDA oversight.
As part of the FDA’s fiscal year 2023 budget request, the agency has requested additional authorities to expand its ability to use remote regulatory tools across all commodities. The agency currently relies on voluntary cooperation for the use of these tools for non-drug establishments or when the RRA does not involve assessing a food importer’s compliance with Foreign Supplier Verification Programs regulations.