

Industry Feedback: FDA’s Proposed Quality Metrics Program

The FDA gained the input of bio/pharma companies, CDMOs/CMOs, and industry groups on its proposed Quality Metrics Reporting Program, which will gather info from manufacturers and be used by the FDA in its quality surveillance activities. What did the industry have to say?
FDA’s proposed Quality Metrics Reporting Program
After a multi-year process to get a Quality Metrics Reporting Program into place, the US Food and Drug Administration offered a new direction for the program and sought the input from bio/pharmaceutical companies, CDMOs/CMOs, industry groups, and other stakeholders on its proposal. The comment period for that input ended earlier this month (June 2022), so what did the industry have to say?
The FDA’s impetus for a Quality Metric Reporting Program is to gather information from manufacturers to be used by the FDA in its quality surveillance activities. Quality metrics are objective means of measuring, evaluating, and monitoring the product and process lifecycle to proactively identify and mitigate quality risks, thereby managing operations at higher levels of safety, efficacy, delivery, and performance as well as drive continuous improvement in manufacturing. Although current good manufacturing practices (CGMPs) are used in the industry, the FDA explains that cGMPs do not necessarily indicate whether a manufacturer is investing in improvements and striving for sustainable compliance, which is the state of having consistent control over manufacturing performance and quality.
Quality metrics data, therefore, play a crucial role in evaluating the effectiveness of a pharmaceutical quality system and to achieve sustainable cGMP compliance and supply-chain robustness. Quality metrics also play an important role in supplier oversight and can be used to inform the oversight of outsourced activities and material suppliers as well as appropriate monitoring activities to minimize supply-chain disruptions. Quality metrics data provided by drug manufactures can also assist the FDA in developing compliance and inspection policies and practices to improve the agency’s ability to predict, and therefore possibly mitigate, future drug shortages, and to encourage innovative quality management systems for pharmaceutical manufacturing. For example, quality metrics data can be applied to FDA’s risk-based inspection scheduling, reduce the frequency and/or length of routine surveillance inspections, and provide ongoing insight into manufacturing operations between inspections.
But what those quality metrics should be, how are they defined, how that data/information is gathered by manufacturers, and the reporting requirements to the FDA has been a source of much debate and discussion in the industry.
In what became a multiyear process, in March (March 2022), the FDA refined its proposed Quality Metrics Reporting Program based on lessons learned from two pilot programs with industry, a Site Visit Program, and a Quality Metrics Feedback Program, as well as stakeholder feedback on FDA’s 2016 revised draft guidance, Submission of Quality Metrics Data.
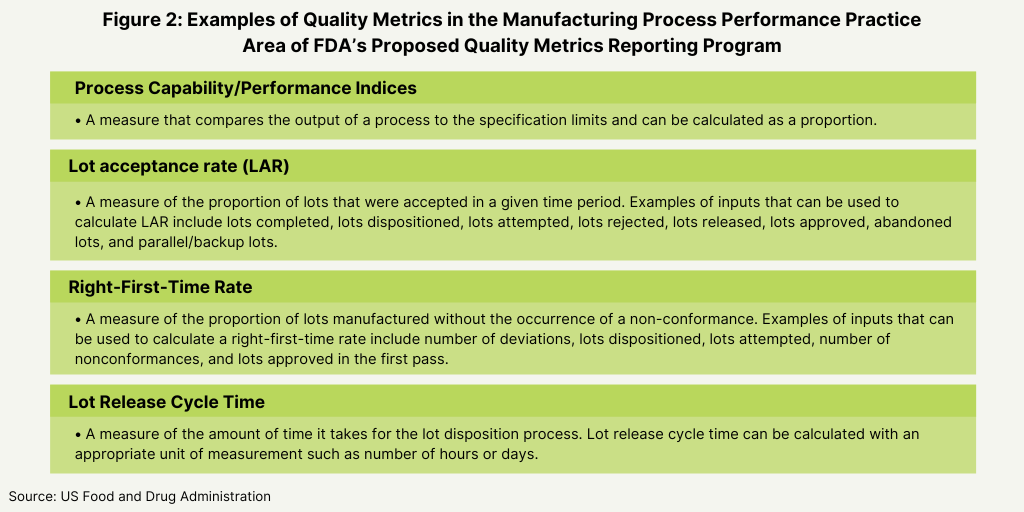
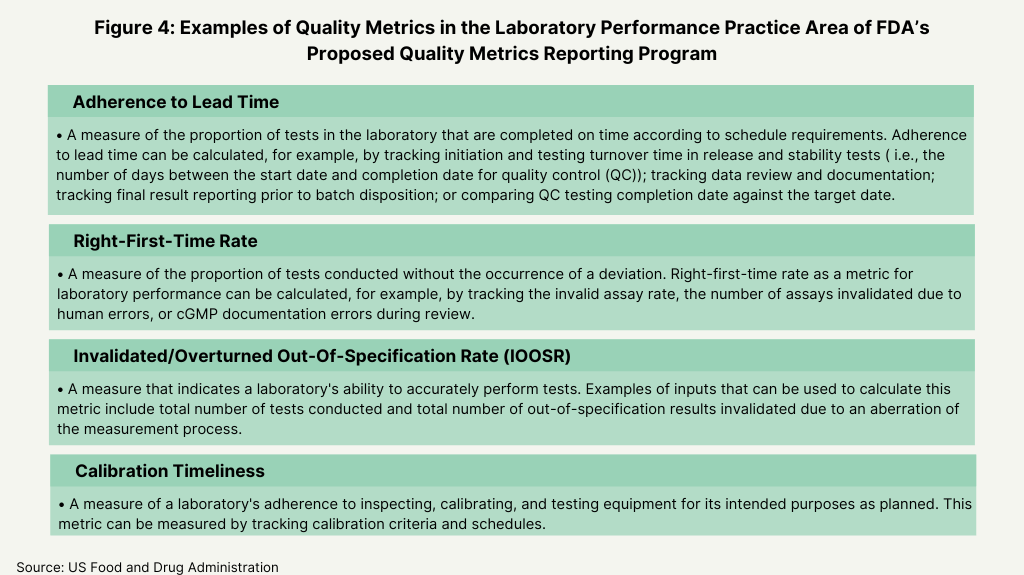
In its proposed plan, the FDA identified four main areas as the basis for the metrics for its program: manufacturing process performance, pharmaceutical quality system effectiveness, laboratory performance, and supply-chain robustness (see Figure 1 below). Its proposal defines these four practice areas and the individual metrics that will fit into each area as a basis of reporting (see Figures 2-5 below).

Industry feedback
To gain the feedback from the industry, the agency posed a series of questions to gain specific input of the feasibility and utility of these metrics. With respect to reporting levels, the FDA wanted to know if reporting should be aggregated at an establishment (i.e., facility level) or product level, and if reporting at an establishment level would facilitate submission of quality metrics data by CMOs. The FDA also wanted to know what other practice areas other than the four general practice areas (see Figure 1 above) should be considered, and if the examples of the quality metrics proposed by the FDA are an appropriate measure for each designated practice area (see Figures 2-5 below) and what other metrics should the FDA consider. The FDA also wanted to know if there were considerations unique to specific product categories (e.g., generic drug products, over-the-counter drug products, or biological products) that should be addressed in the Quality Metrics Reporting Program and what the optimal reporting frequency for quality metrics data submissions (e.g., monthly, quarterly, or yearly, and segmented by quarter or month) should be. The agency also asked for feedback how to handle data from both domestic and foreign establishments, and for feedback on any other aspects of the its proposed direction for the program that the FDA should address in future policy.
In all, the FDA received feedback from 33 industry groups, bio/pharmaceutical companies, CDMOs/CMOs, and individuals. Feedback included comments from industry associations: the Association for Accessible Medicines (AAM), which represents manufacturers and distributors of finished generic pharmaceutical products, bulk active pharmaceutical chemicals, and suppliers of other goods and services to the generic pharmaceutical industry; the Parenteral Drug Association (PDA), a professional association of more than 10,000 individual scientists involved in pharmaceutical, biological, and device manufacturing and quality; the Active Pharmaceutical Ingredients Committee (APIC) of the European Chemical Industry Council (CEFIC), which represents European chemical manufacturers; the Pharma & Biopharma Outsourcing Association (PBOA), an association representing CDMOs and CMOs; the International Society for Pharmaceutical Engineering (ISPE), an organization of individual members from bio/pharmaceutical companies, CMOs, suppliers and service providers focused on scientific, technical, and regulatory issues; and the Biosimilars Forum, which represents companies with the most significant US biosimilars development portfolios Also providing input were bio/pharmaceutical companies, including AstraZeneca, Bristol-Myers Squibb, Merck KGaA, and Pfizer, and CDMOs/CMOs, including Lonza and Catalent.

As might be expected, there was no clear consensus among those providing feedback on the overall redirection of FDA’s proposed Quality Metrics Reporting Program and on the particular questions in which the FDA was seeking industry input. Concerns by those providing feedback relate to the overall focus, feasibility, usefulness, and the value of the reporting program as a whole, given the amount of data already submitted to the FDA and the additional burden and feasibility of collecting and reporting additional data and the utility of that data. Part of the FDA’s plan is to allow manufacturers flexibility in reporting their own metrics under the four practice areas proposed by the FDA (manufacturing process performance, pharmaceutical quality system effectiveness, laboratory performance, and supply-chain robustness). While the industry agreed with providing flexibility, some were concerned that allowing for individual metrics reporting would not achieve the intended goal of common industry quality metrics and that the four proposed practice areas could be further refined. There was also mixed opinion as to how CDMOs/CMOs should participate in the program, given some unique considerations of CDMOs/CMOs, including their ability to collect and report data with sponsor companies “owning” that data. Some highlights of that feedback from industry groups are outlined below.
Association for Accessible Medicines (AAM). The Association for Accessible Medicines (AAM), which represents the generic-drug manufacturers and their suppliers, raised concerns about the new direction that the FDA is taking in its proposed Quality Metrics Reporting Program. Chief among those concerns is a lack of statutory authority by the FDA to establish a Quality Metrics Reporting Program in which participation or submission of data is compulsory or in which information about participation is made public.
In addition, the AAM raised concerns that the proposed program may incorporate metrics that have not been validated and that may not be relevant to quality. Although it acknowledged that increased flexibility to manufacturers is useful, it pointed to the difficulty in coming to standardized reporting if the FDA allows firms to submit unique metrics and to use that data as the basis of reporting and that a set of specific validated and industry-recognized metrics across all participating firms would have greater value. It added that if each individual firm decides which metrics to compile and submit to the FDA, it would provide incentives to manufacturers to choose metrics that would present their firm and establishment in the best possible light rather than choose metrics that more meaningfully reflect the firm and facility’s quality performance and that such metrics could also result in submission to the FDA of metrics of limited or no utility to the FDA.
Instead, the AAM said outlined five main principles relating to a Quality Metrics Reporting Program: (1) the program must be within the bounds of clear statutory authority; (2) quality metrics data should be collectible and participation must be feasible; (3) quality metrics should be validated and correlated to FDA’s goals; (4) quality metrics should be normalized by product type and product risk; (5) quality metrics should not create unintended incentives.
In addition, the AAM agreed that should be aggregated at an establishment level (i.e., meaning facility level) as opposed to being aggregated at the product level, an issue of particular importance for generic-drug companies, given the multitude of products that they manufacture at both internal and external sites. The AAM also said that covered establishments should not be responsible for submitting information to FDA from contract manufacturers, but instead metrics for contract manufacturers should be reported to FDA by the contract manufacturers themselves.
Pharma & Biopharma Outsourcing Association (PBOA). The Pharmaceutical & Biopharmaceutical Outsourcing Association (PBOA), which represents CDMOs/CMOs, raised concerns on the overall focus and direction of FDA’s proposed Quality Metrics Reporting Program and to the value of adding further information over what manufacturers currently provide to the FDA. “Our members are very concerned that collecting data beyond the wealth of information currently in FDA possession without clarity of the end goal will result in a great deal of effort on the part of industry to assemble the data and even more effort on FDA’s part to parse and interpret the data with very little likelihood of meaningful benefit, “said the PBOA in its comments of June 6, 2022 to the FDA.
In addition, PBOA said it is also concerned that the metrics collected will vary dramatically depending on site versus product metrics, the development status of drugs being measured, annual manufacturing demand, and the age of the manufacturing product/process. “All of these factors, and many more, will overshadow how many of these metrics are interpreted,” it said in its comments. Among its suggestions, the PBOA recommended restricting collection of metrics exclusively to commercial products and excluding clinical/development products from the scope of reporting activity.
PBOA also further recommended that the FDA take into consideration whether a site is a CDMO site or a non-CDMO site in assessing the quality metrics data from that site. “In most cases, CDMOs manufacture a wide range of products and in some cases very complex products; these are both scenarios that could likely cause a CDMO site to ‘score’ lower on metrics than, for example, an innovator’s site dedicated to a single product,” said the PBOA in its comments.
PBOA is also strongly recommending the agency take into account the timelines for existing reporting requirements, so as not to unduly burden industry with a surge of reporting data, particularly in light of that CDMOs/CMOs do not “own” much of the data that FDA wants sites to report, rather sponsor companies do. It says more reporting requirements require time to compile, review, and interpret data, draft reports, and collect approval from the owners of this data, and FDA must take these burdens into consideration and clarify its goals for such reporting.
PBOA also pointed to unique issues with CDMOs/CMOs that are important for the FDA to consider in its Quality Metrics Reporting Program. It noted that implementing reporting requirements would necessitate that a CDMO update every quality agreement with its clients to define roles/responsibilities regarding metrics reporting and that sponsor companies would have varied preferences in defining those responsibilities. It also noted that poor metrics may not be indicative of a poor quality mindset of a site but rather relate to the manufacturing process itself. It noted that older products or products with more complex manufacturing processes may not score well due to the manufacturing process itself and that a CDMO/CMO has limited ability to change or update a manufacturing process without prior sponsor approval and would require filings to multiple regulatory regimes.

Active Pharmaceutical Ingredients Committee (APIC) of the European Chemical Industry Council (CEFIC). which represents European chemical manufacturers. The Active Pharmaceutical Ingredients Committee (APIC) of the European Chemical Industry Council (CEFIC), which represents European chemical manufacturers, commented that the metrics proposed in the FDA’s proposed plan have a too wide field of application. It pointed to metrics, such overall equipment effectiveness (see Figure 3 above) or on time in full (see Figure 5 above) are not parameters strictly related to GMP, and therefore are not so easy to be collected and verified by a company’s quality unit. APIC suggested to start the submission to FDA of metrics with strict related quality impact, as FDA had recommended in an earlier proposal. It also said that FDA’s decision to leave the definition of quality metrics flexible, while enabling all categories of facilities to provide useful reporting, would provide great variability between establishments.
Parenteral Drug Association (PDA). The Parenteral Drug Association (PDA) emphasized the importance that the Quality Metrics Reporting Program for achieving continuous improvement in quality rather than emphasizing the scores from such a program. “To be effective and successful, PDA believes that the FDA
Metrics Program should avoid becoming a strict compliance requirement (for example inspectors arriving at a site with a metrics report in hand) as that would likely lead to unintended consequences and behaviors that are counterproductive to developing a good quality culture and continuous improvement mindset,” said the PDA in its comments of June 7, 2022 to the FDA. “This may occur especially at sites with less mature quality systems that might overemphasize having ‘good’ metrics results rather than focusing on learning from the metrics to further continuous improvement of the underlying systems and processes. Periods of time where there is an increase in variability of specific metrics will occur for even the best performing site for reasons that must be considered when evaluating the data.”
PDA also said in its comments that while the FDA’s proposal provides a better understanding of the FDA’s current view, the industry still has several questions where additional detail would be helpful. Some of those questions include: (1) how will the metrics data be used internally at FDA; (2) what type of connection is foreseen between the Quality Metrics Program and other FDA projects: the proposed Quality Management Maturity Rating System, the Risk-Based Inspection Model, and the Remote Assessment Program; and (3) what type of communication should a site expect from FDA after the submission of metrics data; and (4) Will FDA be compiling the data for all products from a CMO site and then analyzing the metrics across products for that CMO.




