
Bio/Pharma Outlook 2026: The Year Ahead

What are the key issues on the bio/pharmaceutical industry’s radar in 2026? DCAT Value Chain Insights weighs in on the key trends and developments shaping the industry in the year ahead: from manufacturing and supply lines to product innovation and new drug development.
By Patricia Van Arnum, Editorial Director, DCAT, pvanarnum@dcat.org
US-based tariffs: what may be next
As we begin 2026, still high on the industry’s radar is evolving US tariff/trade policy. Last year (2025) was a year of great change with the Trump Administration moving forward with plans to impose reciprocal tariffs, country-by-country-based tariffs designed to counter trade imbalances with US trading partners, and as a means to improve US competitiveness, including in manufacturing. Those reciprocal tariffs took an up-and-down path in terms of timelines for implementation and the actual tariff rates. The reciprocal tariffs were first scheduled to go into effect on April 9, 2025, but the Administration placed a 90-day pause (until July 9, 2025) on their implementation to enable countries to negotiate these tariffs with the US government. The US government then extended the deadline again to August 7, 2025, except for certain countries, which were sent letters from the White House that specified their tariffs, as the US government continued to negotiate individual deals with its trading partners. Since that time, the US has struck deals/framework agreements with key trading partners of particular relevance to the pharmaceutical industry, including the European Union (EU) the UK, Switzerland, Japan, Canada, China, and India.

Now in 2026, several issues loom large. Top of mind in the tariff battle is a pending decision by the US Supreme Court on the legality of the Administration to impose reciprocal tariffs under executive action. The US Supreme Court heard oral arguments in November (November 2025) in two cases to decide whether the current Administration has the legal authority to impose tariffs under the International Emergency Economic Powers Act (IEEPA), which was used to impose reciprocal tariffs on a country-by-country basis. Hearing oral arguments is only the first step in the US Supreme Court process as it reviews and considers these cases. Until a decision is reached, reciprocal tariffs remain in place. As of press time (January 8, 2026), indications were that the US Supreme Court may be issuing a ruling shortly or announcing when it planned to do so although no official announcement has been thus far made.

The prospect of another form of US tariffs, specifically imposed on the pharmaceutical industry, also looms large in 2026, so-called Section 232 tariffs. Section 232 of the Trade Expansion Act of 1962, as amended, allows the President to impose import restrictions based on an investigation and affirmative determination by the US Department of Commerce that certain imports threaten to impair US national security. The Trump Administration has used this authority to impose tariffs on other industries, and the process for evaluating potentially imposing such tariffs on the pharmaceutical industry was initiated last April (April 2025). Throughout 2025, that evaluative process and signals by the Trump Administration that it was considering pharmaceutical-industry specific tariffs continued—but to date (as of January 8, 2026), no such tariffs have been imposed on the industry as a whole.
In the meantime, the Trump Administration has struck deals with individual pharmaceutical companies to provide a three-year exemption for Section 252 tariffs in return for commitments for increasing capital investment in the US and agreeing to reduce drug prices for certain products (as outlined in individual company agreements) to be in line with the Administration’s policy interest for most-favored-nation drug pricing. The premise behind most-favored-nation drug pricing is that the US pays higher prescription drug costs comparative to other developed countries and therefore assumes a larger share of the costs of drugs and that measures should be taken to reduce the differential in the prices of prescription drugs in the US compared to other developed countries, where prescription drug prices are lower. The individual company agreements reached with the Administration to date include those with Amgen, AstraZeneca, Bristol Myers Squibb, Boehringer Ingelheim, Eli Lilly and Company, Roche’s Genentech, Gilead Sciences, GSK, Johnson & Johnson, Merck & Co., Novo Nordisk, Novartis, Pfizer, and Sanofi.
US-based bio/pharmaceutical manufacturing
Hand-in-hand with US tariff/trade policy is increased capital investment in the US by pharmaceutical companies. Part of the policy rationale for imposing tariffs is to increase US competitiveness in manufacturing by creating incentives for companies to base operations in the US to avoid tariff burden. Last year (2025), the industry saw a slate of announced expansions in the US—greenfield projects and expansions of existing facilities for manufacturing, research and development, and other capital projects—by the large bio/pharmaceutical companies. These multi-year investments included announcements made by AbbVie ($10 billion), Amgen ($3 billion), AstraZeneca ($50 billion), Biogen ($2 billion), Eli Lilly and Company ($50 billion), Gilead Sciences ($32 billion), GSK ($30 billion), Johnson & Johnson ($55 billion), Merck & Co. ($70 billion), Novartis ($23 billion), Novo Nordisk ($4.1 billion), Pfizer ($70 billion), Regeneron Pharmaceuticals ($2 billion), Roche ($50 billion), and Sanofi ($20 billion). This year (2026) is expected to see further details on specific projects and raises an important question for the industry as to whether these investments constitute overall geographic shifts to US-based manufacturing.
The EU: The Critical Medicines Act
Just as the US is seeking to increase domestic manufacturing through public policy so is the EU by proposing measures to reduce shortages of essential/critical medicines by improving security of supply and availability in the EU. In March (March 2025), the European Commission sought to address these issues by publishing a proposal for the Critical Medicines Act, a new EU regulation that aims to improve the availability and security of supply of essential medicines within the EU. The proposal seeks to make Europe’s healthcare system more resilient and self-sufficient to better withstand global health challenges that threaten access to essential medicines, such as pandemics, natural disasters, and supply chain disruptions. The new rules would complement the proposals to revise the EU’s pharmaceutical legislation (see “Europe at a crossroads later in the article), which also contain provisions on managing shortages and the supply of medicinal products.
As part of the legislative process to enact the Critical Medicines Act, last month (December 2025), the Council of the European Union agreed to its position on the Critical Medicines Act, which provide new rules to incentivize supply chain diversification, facilitate collaborative procurement models, and create incentives to boost pharmaceutical manufacturing in EU countries. Once the European Parliament has established its position, the two institutions will enter into negotiations to agree on the final text. Specifically, the Critical Medicines Act aims to improve the availability, security, and resilience of the supply chain for essential medicines by: (1) diversifying the supply chain to reduce dependence on a limited number of suppliers; (2) providing incentives for collaborative procurement initiatives to help EU member states purchase medicines more effectively and reduce costs; and (3) boosting EU pharmaceutical manufacturing to ensure that critical medicines are produced within the EU by reducing reliance on third countries (i.e., non-EU countries). Key for 2026 is the progression of the proposed legislation to formal approval in the EU.
Product innovation: how will the industry fare in 2026?
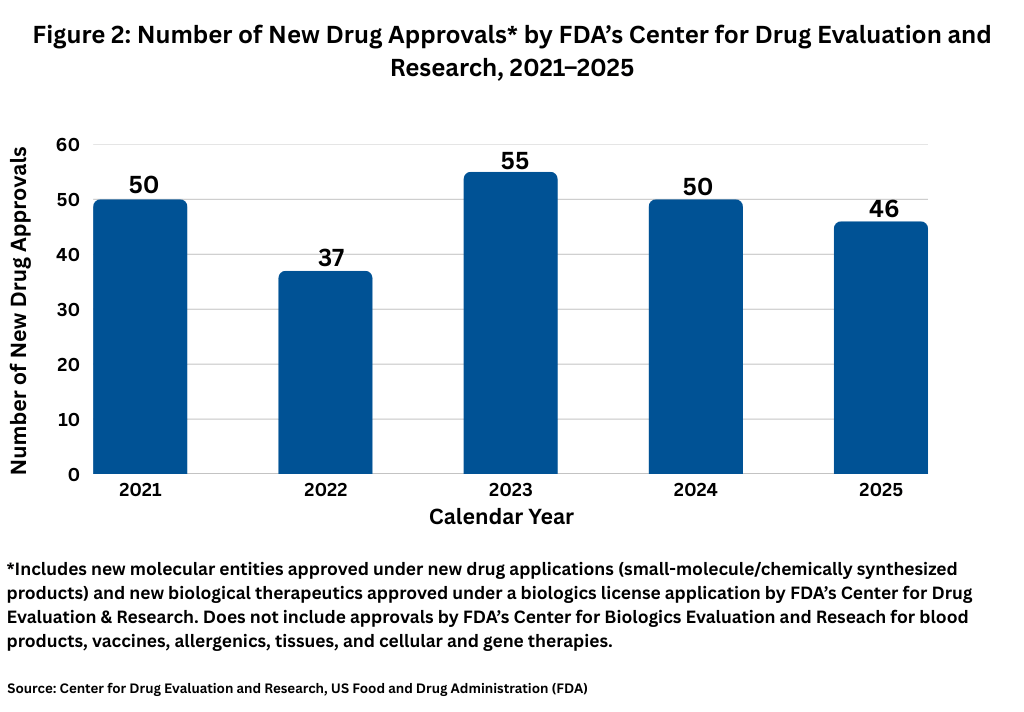
Product innovation is central to the health of the bio/pharmaceutical industry, and the number of new drug approvals by the US Food and Drug Administration (FDA) is an important barometer of the level of that innovation. Last year (2025) presented some distinct challenges on the regulatory front. First, staffing and budgetary cuts at the agency, changes in FDA leadership, and a seven-week shutdown of the US government in which reviews of new drug filings were limited made 2025 a different environment. As the industry enters 2026, a key question is did these changes impact new drug approvals in 2025 and what may be in store for 2026?
Although new drug approvals by FDA’s Center for Drug Evaluation and Research (CDER) were somewhat down in 2025, they remained largely in line with previous approval levels. In 2025, FDA’s CDER approved 46 new molecular entities approved under new drug applications (small-molecule/chemically synthesized products) and new biological therapeutics, such as recombinant proteins and monoclonal antibodies, approved under a biologics license application. Other biologic-based products, including blood products, vaccines, allergenics, tissues, and cellular and gene therapies, are reviewed and approved by a separate center within FDA, the Center for Biologics Evaluation and Research (CBER), and are not part of this analysis. Figure 2 shows full-year drug approvals from 2021 to 2025, and with the exception of 2022 (which had 37 new drug approvals) and 2025 (which had 46 new drug approvals), there have been 50 or more new drug approvals in a given year over the past five-year period.
Europe at a crossroads
Also high on the industry’s radar in 2026 will be the progression of major policy reforms in the EU being taken to boost competitiveness, innovation, and security of medicines supply. Last month (December 2026), EU policymakers took a significant step in advancing major reforms to the EU’s regulatory framework, representing the most significant overhaul of the regulatory framework of pharmaceuticals in the EU in over two decades. The reforms to the EU’s pharmaceutical legislation was first proposed by the European Commission in April 2023. The European Parliament adopted its position in April 2024, and the Council of the European Union in June 2025. A political agreement reached last month (December 2025) is now subject to formal approval by the European Parliament and the Council of the European Union.
Key for both innovator and generics/biosimilars companies are proposed measures for data protection exclusivity. The proposed measures set a regulatory data protection period (during which other companies cannot access product data) of eight years, with one additional year of market protection (during which generic or biosimilar products cannot be sold) following a marketing authorization. Pharmaceutical companies would be eligible for additional periods of market protection under certain conditions. If the particular product addresses an unmet medical need, an additional 12 months of exclusivity would be granted. Also, an additional 12 months of exclusivity would be granted if the product contains a new active substance and meets a combination of conditions on comparative clinical trials, clinical trials carried out in several EU member states, and the obligation to apply for market authorization within 90 days after the submission of the application for the first marketing authorization outside the Union. In addition, if a company obtains an authorization for one or more new therapeutic indications that bring a significant clinical benefit in comparison with existing therapies, an additional 12 months of exclusivity would be granted. In addition, the proposed measures envisage a cap of 11 years on combined regulatory protection period. Orphan medicinal products addressing a disease with no current available medicinal treatment (“breakthrough orphan medicinal products”) would receive up to 11 years of market exclusivity.
To support earlier market entry of generic and biosimilar medicinal products, the deal clarifies the scope of the “Bolar” exemption (which allows manufacturers to conduct certain activities during the market protection period of the original product). The proposed measures specify that patent rights would not be infringed when necessary studies, trials, and other activities are conducted for the purposes of obtaining marketing authorizations, conducting health technology assessments, obtaining pricing and reimbursement approvals, or submitting procurement tender applications.
Industry feedback
The question for 2026 and going forward is whether the EU pharma legislation, once formally enacted, would achieve the desired policy goal of greater product innovation and improved competitiveness for the European pharmaceutical industry. The European Federation of Pharmaceutical Industries and Associations (EFPIA), which represents the innovator drug industry in Europe, says while the proposed legislation is a step forward, more need to be done. “Our region has lost a quarter of its global share of investment to other parts of the world in two decades while our share of clinical trials has halved,” said Nathalie Moll, Director General, EFPIA, in a December 11, 2025, statement, in commenting on the EU pharma legislative package. “If this is the legislative framework that is expected to attract the medicines innovation of the next 20 years to Europe, the outcome of the trialogues is underwhelming. If Europe truly wants to be competitive, it needs to increase investment in innovative medicines, strengthen rather than weaken IP [intellectual property] and make the process of getting new medicines to patients faster and more connected. We now look to the Biotech Act and remain hopeful of policies to reverse the current trends, as well as a renewed enthusiasm to value and prioritize healthcare innovation in Europe.”
For the generics/biosimilars industry, feedback to the proposed legislation has been broadly positive although Medicines for Europe, which represents the generics/biosimilars industry in Europe, points to additional changes it deems important. “The deal agreed between Parliament and Council on the general pharmaceutical legislation review is an important step forward for the European pharmaceutical industry at a time of great geopolitical uncertainty,” said Medicines for Europe, in a December 11, 2025, statement. “Medicines for Europe has supported the objectives of the reform from the start: improving equity of access for all Europeans, ensuring timely access to generic, biosimilar and value-added medicines, and modernizing the regulatory framework through more digitalization, harmonization and optimization.”
However, Medicines to Europe points to additional reforms it support. With respect to equitable access, the association says: “[W]e would have supported a more ambitious reform for better access to medicines in Central and Eastern European and smaller member states and will work with the EU to play our role in filling the access gaps as much as possible,” it said in its statement. With respect to timely access of medicines, Medicines for Europe supports the Bolar harmonization. With respect to digitalization and harmonization of pharmaceutical regulation, the association says the the legislation “will align the pharmaceutical legislation with the objectives of the EMA [European Medicines Agency] extended mandate legislation to prevent and mitigate medicine shortages although longer shortage notification periods will increase shortages ‘false alerts’ and overload national competent authorities.”
Other proposed measures of the EU pharma legislative package
The proposed reforms establish several new rules for marketing authorization holders and public health authorities to cooperate to ensure continuity of supply and availability of critical medicines in the EU. Companies holding marketing authorizations for medicinal products would be required to put in place and update shortage prevention plans for medicinal products identified by the European Commission. Expected and actual drug shortages would be monitored at both national and EU levels, based on notifications from marketing authorization holders. The proposed measures also call for the establishment of an EU list of critical medicines for which supply-chain vulnerability assessments will be required to be carried out by pharmaceutical companies. The Critical Medicines Act is a further legislative vehicle to address drug shortages and assure security of supply in the EU.
In addition, under the proposed reforms under the EU pharma legislation, the European Medicines Agency (EMA), the EU regulatory body for pharmaceuticals, would put into place several measures to streamline the drug review process. The updated rules would simplify EMA’s internal functioning to enable it to treat market authorization requests more rapidly. Marketing authorization applications would be submitted electronically in a common format. Marketing authorization for a medicinal product would be valid by default for an unlimited period, avoiding the unnecessary administrative burden linked to renewals (the EMA would still have the possibility to limit validity, on safety grounds). Under special conditions, the European Commission may set up regulatory sandboxes, under the direct supervision of the competent authorities, to allow the development and testing of new and innovative therapies that cannot by developed under current rules,. Such a regulatory sandbox would be established at the suggestion of EMA and in consultation with EU member states.
Also, under the proposed measures, EMA would simplify its committee structure from five to two scientific committees, the Committee for Human Medicinal Products and the Pharmacovigilance Risk Assessment Committee. The streamlined structure is intended to facilitate reduced assessment times from 210 to 180 days and free up scientific resources to strengthen pre-authorization support to medicine developers.
The proposed measures also support the creation of adapted frameworks for certain non-standard categories of medicines, such as personalized therapies, and provide further support for pediatric drug development. The proposed measures specify improvements in the efficiency of the process for studying medicines in children, including formalizing a new process for iterative pediatric investigation plans.
The proposed legislation also strengthens provisions relating to environmental risk assessment, including special methodologies to evaluate the risk for antimicrobial resistance selection. Among new measures to promote the prudent use of antimicrobials, the proposed legislation introduces stricter requirements, such as compulsory medical prescriptions for all antimicrobials, specific information requirements to be provided with the package leaflet, and an “awareness card” in paper format in case the leaflet is made available only electronically. The proposed legislation also includes measures to encourage the development of antimicrobials by introducing a “transferable data exclusivity voucher” for priority antimicrobials, giving the right to 12 additional months of data protection for one authorized product.
Next steps in the EU
The provisional agreement for the EU pharma legislation is now subject to formal approval by the European Parliament and the Council of the European Union. Once the text of the proposed EU pharmaceutical legislation is formally approved, over the coming months and years (as reported on December 11, 2025), EMA will work together with the European Commission and EU member states to develop relevant guidance for applicants and marketing authorization holders to comply with the new legal framework. To ensure the smooth implementation of the legislation, EMA says its stakeholders will be kept informed and actively involved in the implementation process on specific technical and procedural aspects of the reform. EMA plans to publish a new web page that will serve as a gateway and central repository of information on implementation of the new legislation. The webpage will be updated as implementation work progresses and guidance for pharmaceutical companies becomes available.




