FDA, GMP Inspections and COVID-19: What’s Next?

The FDA cut back inspections in 2020 due to COVID-19 and reverted to remote inspections and alternative tools, but what has been the impact? The FDA conducted 52 domestic inspections from March to September 2020, compared to about 400 in prior years, and only three foreign inspections. A government watchdog agency is recommending changes. What are they?
FDA inspections and COVID-19
One of the consequences of the COVID-19 pandemic has been a significant reduction in the number of inspections conducted by the US Food and Drug Administration (FDA). Although the FDA instituted alternative methods during the COVID-19 pandemic to replace onsite, in-person inspections, a recent report by the Government Accountability Office (GAO), a legislative branch government agency that provides auditing, evaluation, and investigative services for the US Congress, examined the extent of inspection reductions, the impact, and recommendations to improve.
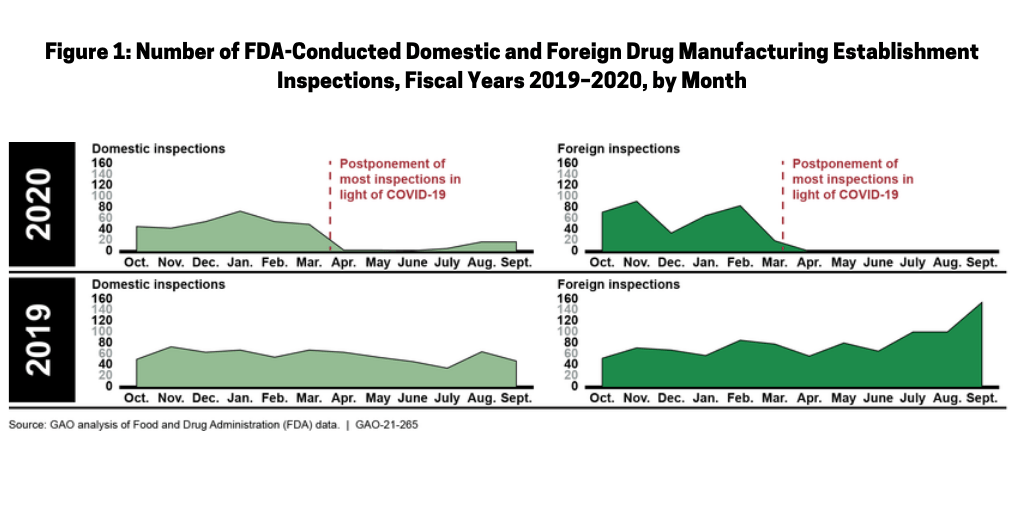
Prior to COVID-19, the FDA typically conducted more than 1,600 inspections of foreign and domestic drug manufacturing establishments each year, but inspections have been reduced significantly, according to the GAO report. The total number of FDA inspections of foreign and domestic establishments was 56% lower in fiscal year 2020 than during each of the previous two fiscal years. Figure 1 shows the reduction in inspections pre- and post-COVID-19, respectively in fiscal years 2019 and 2020.
Domestic inspections pre- and post-COVID-19
The number of FDA domestic inspections (i.e., those conducted in the US) fell significantly as a result of COVID-19. From March to October 1, 2020 (the latest period in which data were available for the GAO analysis released in January 2021), the FDA conducted 52 domestic inspections, compared to about 400 inspections conducted during this time period in the two previous years. In mid-March 2020, the FDA limited domestic inspections to only those deemed “mission critical.” In July 2020, the FDA announced that it planned to resume domestic inspections, contingent on a rating system that incorporates information on COVID-19 infection trends in a geographic area. Depending on an area’s rating, the FDA’s inspection activities were to range from “mission critical” inspections only, to the resumption of all inspections. According to the FDA’s area rating data, as of December 3, 2020, conditions were appropriate for conducting routine surveillance inspections in 49 US counties, with regulatory activity limited to mission critical inspections only in the more than 3,000 remaining counties. The FDA also announced that to help assure the safety of inspection staff and establishment employees, for the foreseeable future, domestic inspections will be preannounced, as has typically been done for most foreign inspections. Previously, almost all domestic inspections were unannounced.
Foreign inspections pre- and post-COVID-19
In early March 2020, the FDA announced that, in light of the COVID-19 pandemic and citing the safety of its employees, the agency would temporarily not conduct any foreign inspections other than those deemed “mission critical.” From the pause in inspections in March to October 1, 2020 (the most recent data available at the time of the GAO analysis), the FDA conducted three foreign mission critical inspections. In contrast, from March to September of each of the prior two years, the FDA conducted more than 600 foreign inspections. As of January 2021, the FDA had not set a date for resuming regular foreign surveillance inspections in all countries, but said it continues to monitor the global situation and remains in contact with foreign regulators to inform the FDA’s assessment of the feasibility of returning to foreign surveillance inspections as conditions improve. For example, according to the FDA, in January 2021, staff in the agency’s China office had begun conducting surveillance inspections in China and staff in the agency’s India office would begin conducting surveillance inspections in India shortly, according to the GAO report.
Alternative tools for inspections used by the FDA
During COVID-19, the FDA has been using alternative tools for inspections, but the GAO cautions that these tools “are not a comprehensive or long-term substitute for inspections.” The FDA has used alternative tools for oversight of drug manufacturing quality while inspections have been paused; these alternative tools include inspections conducted by foreign regulators, requesting and reviewing records and other information, and sampling and testing. “Though FDA has determined that inspections conducted by certain European regulators are equivalent to an FDA inspection, other tools provide useful information, but are not equivalent to an FDA inspection,” says the GAO in its report.
In light of the pause in inspections due to COVID-19, the FDA established a policy expanding the use of the mutual recognition agreement it has with the European Union to include inspections conducted outside of Europe by European regulators. The FDA also began using information from inspections conducted by other regulators—specifically, regulators that are among the 53 members of the Pharmaceutical Inspection Cooperation Scheme (PIC/S), such as Australia, Canada, Japan, and South Africa—though the agency has not determined that inspection reports from such countries are equivalent to FDA inspections, according to the GAO report. According to FDA officials, in fiscal year 2020, the FDA substituted European regulator inspection reports for over 160 FDA inspections in Europe and requested over 30 additional reports from inspections conducted by European regulators or PIC/S members in China, India, Korea, and Japan, among other countries.
The FDA had previously determined that European regulators had the capability of conducting inspections in Europe that were equivalent to the FDA’s own inspections. In light of the COVID-19 pause in inspections, the FDA also evaluated for equivalence inspections conducted outside Europe by European regulators covered under the mutual recognition agreement. According to FDA officials, as of November 2020, the FDA deemed that inspections conducted outside of Europe from 19 of 28 European regulators can be substituted for an FDA inspection. However, reports for inspections from the other nine European regulators conducted outside of Europe and by PIC/S members can only be used to help obtain “surveillance-level oversight” while inspections are paused and are not full substitutes for an FDA inspection, according to the GAO report. In addition, the FDA’s ability to continue to rely on these tools may be limited, as foreign regulators have also postponed inspections due to COVID-19.
Further, the GAO report noted that many foreign establishments supplying the US market are located where foreign regulator reports may not be available. For example, the GAO report says that FDA officials told GAO officials that, particularly in China and India (two countries that are not PIC/S members), the FDA conducts more foreign inspections than any other regulator. Thus, there may not always be a foreign regulator report to rely on while FDA inspections are paused. In fiscal year 2019, almost half of the FDA’s 977 foreign inspections were in China and India, the two countries which also had the largest number of establishments supplying the US market.
The FDA can request and review records and other information to substitute for FDA inspections in select circumstances. During the COVID-19 pandemic, the FDA substantially increased use of its authority to request that establishments provide records in advance of or in lieu of an inspection, requesting records from establishments in China, India, and the US, among others, according to the GAO report. The FDA can substitute the review of records and other information for conducting a preapproval inspection, but will only do so in certain cases. For example, the FDA may choose to do this if the establishment has an acceptable drug inspection history for related manufacturing operations. The GAO report noted that according to FDA officials, in fiscal year 2020, the FDA made over 130 requests for records and other information to support preapproval applications listing establishments in at least 27 countries.
According to the FDA’s interim policy, the FDA can combine non-European foreign regulator reports with establishment records to support its risk-based surveillance of establishments already manufacturing drugs marketed in the US, but establishment records alone cannot be used as a substitute for an FDA surveillance inspection. The GAO report said that FDA officials told GAO officials that only FDA in-person inspections and European regulator reports can satisfy the agency’s statutory requirements for surveillance inspections. According to FDA officials, in fiscal year 2020, the FDA made over 310 requests for records and other information to support surveillance of establishments in at least 36 countries. If needed, the FDA can request that the establishment provide translated versions of requested records and provide verification that the translation is complete and accurate.
With respect to sampling and testing, the FDA noted in the GAO report that in response to the COVID-19 pandemic, the agency has adjusted its approach to selecting drugs for sampling and testing. Specifically, according to FDA officials as noted in the GAO report, the tool the FDA uses to automatically screen drug imports continues to be adjusted during the COVID-19 pandemic to help the FDA determine where to focus its sampling at the US border. The FDA also has targeted sampling of high-risk and difficult-to-manufacture drugs from foreign establishments that had their inspections postponed. While FDA’s sampling and testing provides the agency with information about a product’s quality attributes, the agency notes that sampling and testing alone do not specifically confirm adherence to quality standards and thus cannot fully replace an FDA inspection. The agency does not test every drug both because the volume of drugs is not feasible to test and only about 1% of the drugs it tests in a given year fail to meet established quality specifications, according to the FDA.
Alternative tools allowed the FDA to take some regulatory action against foreign drug manufacturing establishments with manufacturing deficiencies during the inspection pause. From March 1, 2020 to December 1, 2020, the FDA placed 54 foreign establishments on import alert based on sampling. The FDA has also placed establishments on import alert based on information obtained from other alternative tools, such as a finding of non-compliance in a foreign regulator inspection report or for a foreign establishment refusing to provide records in response to a request from the FDA. According to FDA officials, from March 1, 2020 to December 1, 2020, the FDA placed nine foreign establishments on import alert for refusing FDA records requests or for deficiencies identified during the agency’s review of records provided in response to an FDA records request and one foreign establishment on import alert based on a foreign regulator inspection report.
What’s next
The GAO further examined future challenges by the FDA in its inspection process. “FDA faces future challenges with its preapproval and surveillance oversight activities,” said the GAO in its report. “The agency will face challenges with its drug approval and surveillance activities, particularly given that the alternative tools FDA is using while inspections are postponed are not a long-term or comprehensive substitute.”
Potential backlog. A continued pause in preapproval inspections may lead to future delays in FDA drug approvals. As of November 2020, FDA officials told the GAO that the agency had not experienced a significant impact on approval decisions due to the COVID-19 inspection pause. The FDA notes that it is continuing its work to review and approve drug applications and that, as of October, the agency had approved more than 600 brand name and generic drug applications in 2020. Further, as of November 2020, the FDA reported that it was operating above its 90% on-time action performance goal for approval decisions.
Representatives from three associations representing drug manufacturers stated that, because preapproval inspections may happen months before an application is approved, the postponement of inspections has not had a significant effect on the FDA’s ability to make drug approval decisions yet. However, two of these associations noted that the longer inspections are postponed, the more likely the inability to conduct a preapproval inspection could create larger challenges for the FDA’s ability to make approval decisions. FDA officials said that they are continuing to expand the use of alternative tools to mitigate the effect of the pandemic on the agency’s ability to make approval decisions when inspections are not possible.
The GAO reports that continued postponement of FDA surveillance inspections may result in a backlog, creating the possibility that the FDA may not inspect the establishments prioritized by its risk-based site selection model. As a result of the pause in inspections during the COVID-19 pandemic, the FDA was unable to complete more than 1,000 of its fiscal year 2020 surveillance inspections. The FDA has a strategic goal of shifting toward exclusively risk-driven surveillance inspections. However, the inspection pause has increased the number of mandatory inspections the agency will have to conduct in 2021, threatening that goal, according to the GAO report. According to FDA officials, in order to achieve the agency’s strategic goal of risk-driven surveillance inspections, the FDA seeks to maximize the number of inspections of establishments prioritized by the model. For fiscal years 2019 through 2021, the FDA planned for an annual surveillance inspection capacity of about 1,500 inspections, and it plans a similar inspection capacity for fiscal year 2022.
Recommendations by the GAO
The GAO made several recommendations to improve the inspection process. It said that as inspection plans for future fiscal years are developed, the FDA Commissioner should ensure that such plans identify, analyze, and respond to the issues presented by the backlog of inspections that could jeopardize the goal of risk-driven inspections. The GAO also said that the FDA should fully assess the agency’s alternative inspection tools and consider whether these tools or others could provide the information needed to supplement regular inspection activities or help meet its drug oversight objectives when inspections are not possible in the future.