

API Sourcing: The Supply Side for US-Marketed Drugs

The US is the single largest pharmaceutical market in the world, but it lags behind other countries in API production for drugs marketed in the US. How are the supply lines for APIs evolving for US-marketed drugs?
API supply lines for US market
The US is the largest single pharmaceutical market, accounting for approximately $485 billion, or 38%, of the global pharmaceutical market of $1.205 trillion in 2018, according to information from the IQVIA Institute for Human Data Science. Despite the large market share by the US, the US trails other countries in terms of active pharmaceutical ingredients (APIs) supplied for US-marketed drugs. Last month (October 2019), Janet Woodcock, Director of the Center for Evaluation and Research (CDER) at the US Food and Drug Administration (FDA) testified before the Subcommittee on Health, Committee on Energy and Commerce, US House of Representatives, to provide information on the US position in global pharmaceutical supply chains.
“Historically, the production of medicines for the US population has been domestically based,” said Woodcock in her testimony on October 31, 2019. “However, in recent decades, drug manufacturing has gradually moved out of the United States. This is particularly true for manufacturers of active pharmaceutical ingredients (APIs), the actual drugs that are then formulated into tablets, capsules, injections, etc.”
As of August 2019, 28% of the manufacturing facilities making APIs for US markets were based in the US. The remaining 72% of the API manufacturers supplying the US market were outside the US, which includes 13% in China (see Figure 1). The analysis applies to all FDA-regulated products, which includes prescription drugs (branded and generic), over-the-counter (OTC) drugs, and compounded medications. The FDA’s data show that the number of registered facilities making APIs in China more than doubled between 2010 and 2019.
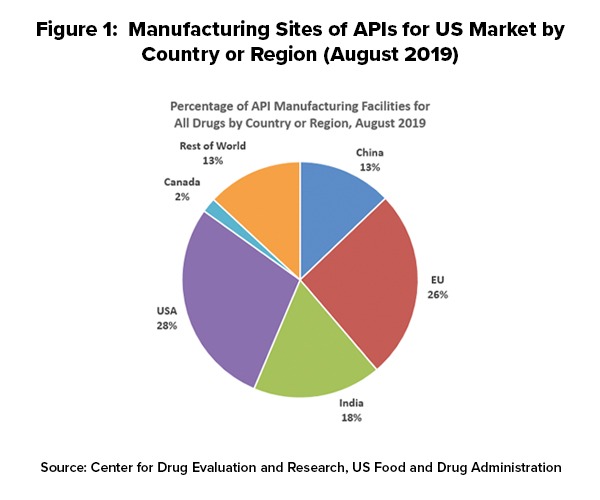
Outside the US, the European Union (EU) represents the largest source of APIs manufactured for US-marketed drugs at 26%, representing the historical and current position of EU-based fine chemical and API manufacturers as well as offshore facilities of pharmaceutical manufacturers. India follows at 18% of APIs supplied to the US and then China at 13% (see Figure 1).
In explaining geographic shifts in API production for US-marketed drugs over the past decade, Woodcock pointed to a variety of factors, including lower-cost labor costs, as a major contributing factor. She cited a 2009 paper by the World Bank, “Exploratory Study on Active Pharmaceutical Ingredient Manufacturing for Essential Medicines,” which specified if a typical Western API company has an average wage index of 100, this index is as low as 8 for a Chinese company and 10 for an Indian company (note: this analysis, published in 2009, does not reflect subsequent cost adjustments or cost equalization since 2009). She also pointed to lower energy costs (electricity and coal) and lower water costs in China. “Chinese firms are also embedded in a network of raw materials and intermediary suppliers, and so have lower shipping and transaction costs for raw materials,” she said. “They also face fewer environmental regulations regarding buying, handling, and disposing of toxic chemicals, leading to lower direct costs for these firms.” She cited a 2011 report by the FDA, “Pathway to Global Product Safety and Quality,” which stated that both China and India have a labor cost advantage and that API manufacturing in India can reduce costs for US and European companies by an estimated 30% to 40%.
Technology-based competitiveness for US-based API supply
Speaking from a US public policy perspective, Woodcock noted that advanced manufacturing technology was an important element in achieving US competitiveness in API supply for US-marketed drugs. “Using traditional pharmaceutical manufacturing technology, a US-based company could never offset the labor and other cost advantages that China enjoys simply by achieving higher productivity,” she said in her testimony. “However, FDA believes that advanced manufacturing technologies could enable US-based pharmaceutical manufacturing to regain its competitiveness with China and other foreign countries, and potentially ensure a stable supply of drugs critical to the health of US patients.”
As outlined by Woodcock, advanced manufacturing is a collective term for new medical-product manufacturing technologies that can improve drug quality, address shortages of medicines, and speed time-to-market. She explained that advanced manufacturing technology, which the FDA supports through its Emerging Technology Program has a smaller facility footprint, lower environmental impact, and more efficient use of human resources than traditional technology, and includes technologies such as continuous manufacturing and 3-D printing.
Drilling down the numbers for API production
 Woodcock provided a further breakdown of the US drug supply and related API production for those products. She provided information from CDER by looking at three main areas: (1) all drugs on the US market, including brand and generic drugs under approved applications, over-the-counter (OTC) drugs, and compounded medications; (2) drugs on the World Health Organization (WHO) Essential Medicines List that are marketed in the US; and for US national security interests, drugs on the medical countermeasures (MCM) lists, which include drugs to counter biological, chemical, nuclear, or radiation threats and influenza.
Woodcock provided a further breakdown of the US drug supply and related API production for those products. She provided information from CDER by looking at three main areas: (1) all drugs on the US market, including brand and generic drugs under approved applications, over-the-counter (OTC) drugs, and compounded medications; (2) drugs on the World Health Organization (WHO) Essential Medicines List that are marketed in the US; and for US national security interests, drugs on the medical countermeasures (MCM) lists, which include drugs to counter biological, chemical, nuclear, or radiation threats and influenza.
She explained that CDER maintains a site catalog of all manufacturing facilities making drugs for the US market, either through an approved application or that have registered and listed to supply drugs for the US market. This includes suppliers for APIs, finished dosage forms (FDF), or both. The APIs manufactured in these facilities may be used in prescription drugs (brand or generic), OTC drugs, and compounded drugs.
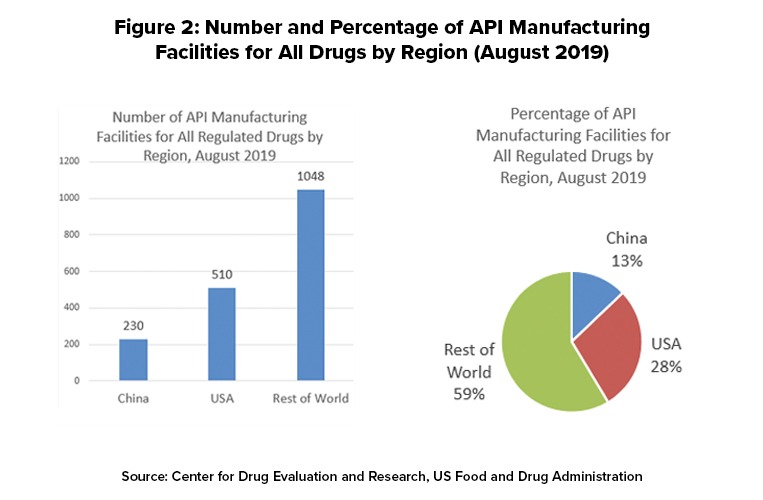
Figure 2 outlines the location of manufacturing facilities used to make APIs for US-marketed drugs. For all regulated drugs (prescription [branded and generics], OTC products, and compounded drugs, the US has 510 manufacturing facilities or 28%, the rest of the world (non-China) 1,048 facilities, or 59%, and China 230 facilities or 13%.
In creating that catalog of manufacturing sites, Woodcock, however, noted several limitations with the data available to CDER, as outlined below.
- Facilities listed in the catalog may or may not be producing APIs. Including a facility in an application or the registration and listing process does not require a facility to produce the API. Producing an API at the facility, or not producing it, is a business decision made by the company.
- Manufacturers are not required to report to FDA whether they are actually producing an API at a facility, and if they are, the volume they are producing.
- APIs made in listed facilities may be used in drugs for both the US and other markets, and some APIs distributed in the US are subsequently formulated into FDF that are then exported.
- Some FDF applications list more than one API supplier in the application. FDA has no visibility into which API supplier an FDF manufacturer uses at any given time.
- CDER has limited information about API suppliers for products that do not need an approved application from FDA to be marketed, such as compounded and OTC monograph drugs. API suppliers for such products may not register their facility with FDA if they are sending material to a drug product manufacturer outside the United States to make the FDF, which is then sold in the United States.
- Information in the catalog is continually being updated. The analysis presented in Woodcock’s testimony is based on August 2019 listings and represents a snapshot at a point in time.
As Woodcock explained, these limitations mean that, although CDER can describe the locations of API manufacturing facilities, it cannot determine with any precision the volume of APIs that given location is actually producing, or the volume of APIs manufactured in a given country that is entering the US market, either directly or indirectly by incorporation into finished dosages manufactured in a given country or other parts of the world.
Essential medicines and US API supply
To provide a public policy view, Woodcock also provided data on the location of API manufacturing facilities used to produce essential medicines for the US market. The World Health Organization (WHO) develops a Model List of Essential Medicines, which serves as a guide for the development of national and institutional essential medicine lists. It is updated and revised every two years by the WHO Expert Committee on Selection and Use of Medicines; the last update was earlier this year (April 2019). The 2019 WHO Essential Medicines List comprises 461 drugs that have been selected by the WHO Expert Committee to meet the most important needs in a health system. This list includes application and non-application products across a wide range of therapeutic categories, such as anesthetic, antibacterial, antidepressant, antiviral, cardiovascular, anti-diabetic, and gastrointestinal agents.
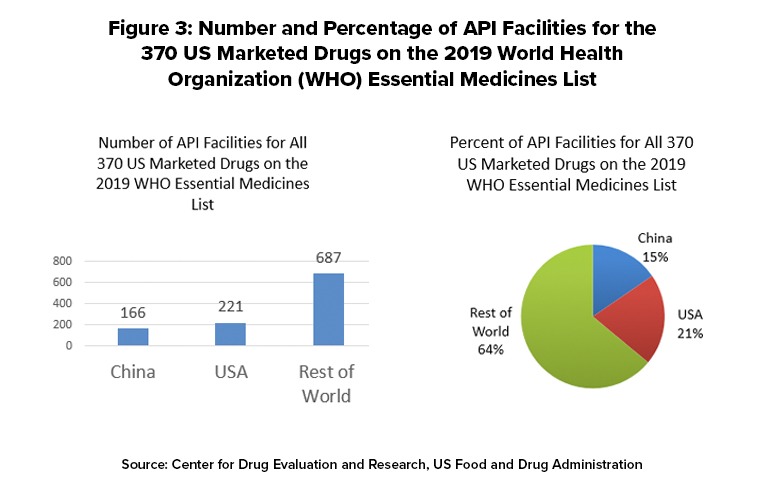
The FDA matched 370 of the drugs on the WHO Essential Medicines List with products listed for the US market and determined the location of the facilities used to make these APIs. The FDA data show that there is a total of 1,079 API facilities worldwide that make the 370 drugs on the WHO list that are marketed in the US Of these, 221 facilities (21%) are in the US, 166 (15%), and 687 (64%) are in the rest of the world (see Figure 3). The FDA determined that there are three WHO essential medicines whose API manufacturers are based only in China. The three medicines are: capreomycin and streptomycin, both indicated to treat Mycobacterium tuberculosis; and sulfadiazine, used to treat chancroid and trachoma, two types of bacterial infection. The distribution of API facilities worldwide varies from drug to drug and may differ from the patterns for all drugs or WHO Essential Medicines List Drugs.
APIs supplied for medical countermeasures
The FDA maintains a list of drugs for the US that are used as medical countermeasures against threats in four categories: biological threats, chemical threats, influenza, and radiation threats. Many of these drugs are contained in strategic drug stockpiles, including the Nation’s Strategic National Stockpile, which can be used in public health emergencies severe enough to cause local supplies to run out. In her testimony, Woodcock outlined the distribution of API facilities making drugs that are used as medical countermeasures.
For APIs for 14 drugs in the biological threat category, the US has 19 API facilities, China has 37 facilities, and the rest of the world has 117. China has six of the facilities producing APIs for the 10 drugs in the chemical threat category, versus 24 in the US and 52 in the rest of the world. China has none of the facilities making APIs for medicines to prevent or treat influenza versus two in the US and 16 in the rest of the world. China also has none of the facilities producing APIs for radiation threats. The US has 13 of these facilities versus 15 in the rest of the world.
Ciprofloxacin and doxycycline are two drugs considered critical as medical countermeasures and used to treat anthrax and plague. The US has one API manufacturing facility for ciprofloxacin, versus three in China, and 21 in other countries. For doxycycline, the US has two API manufacturing facilities, China three, and other countries six.
API supply and US public policy
Woodcock presented the data to Congress as a means to inform public-policy decisions about the security of the US pharmaceutical supply. “The security of the nation’s drug supply rests on three main factors: freedom from dependence on foreign sources of API, the resilience of US market, and the reliability of the facilities that make products for the US market,” she said in her testimony.
With respect to US dependence on non-US sources of APIs, she pointed to probable increased reliance on non-US sources of APIs and current limitations in FDA data to calculate volumes of APIs from different sources for US-marketed drugs.
The number of Chinese facilities producing APIs for the US market has increased over the past decade, as part of a massive movement of pharmaceutical production offshore,” she said in her testimony. “…Absent any intervention, FDA believes that this trend is likely to continue,” she said
However, she pointed out that data available to the FDA do not enable it to calculate the volume of APIs being used for US-marketed drugs from other countries, and what percentage of US drug consumption this represents. Additionally, the FDA data does not specify whether given API facilities are actually producing APIs, how much they are producing, or where the APIs they are producing are being distributed worldwide, including in the US.
Woodcock also addressed how resilient is the U.S. manufacturing base, meaning how quickly could US-based manufacturers increase their production of APIs to meet domestic demand if other countries ceased supplying the US, particularly for drugs on the WHO Essential Medicines list or a subset of those drugs. She said that since the FDA does not currently know whether API manufacturing facilities are actually producing the drug, or in what volume, or what portion of US drug consumption is dependent on APIs from other countries, it cannot perform a reliable gap analysis. To answer these questions, she said the FDA would need to know:
- how much unused capacity exists in the US manufacturing base for APIs;
- how much additional API capacity could supply within a given time period;
- how far this capacity would go in filling the gap between US patients’ needs and the amount available if another country were to reduce or stop the supply to the US market; and
- how long would it take to increase production enough to meet patients’ needs, and whether the financial investment would be sustainable for the pharmaceutical industry.
Advanced manufacturing technologies and other regulatory policy
In her testimony, Woodcock emphasized the importance of the FDA in supporting innovation in pharmaceutical manufacturing technology, such as through advanced manufacturing. The FDA launched the Emerging Technology Program in late 2014 to encourage and support the adoption of innovative technology to modernize pharmaceutical development and manufacturing through close collaboration between the FDA with industry and other relevant stakeholders starting from early technology development.
To reduce barriers to entry for advanced manufacturing, the Emerging Technology Team (ETT) provides a gateway for the early (pre-submission) discussion of innovative technologies and approaches, even before a candidate drug is identified. The ETT supports the entry, assessment, and lifecycle management of advanced manufacturing at CDER. It provides subject-matter experts and fosters coordination within CDER and the FDA’s Office of Regulatory Affairs (ORA) for precedent-setting issues regarding quality and good manufacturing practices. ETT serves as a hub for identification of application-driven regulatory and research needs and provides strategic input for supporting advanced manufacturing innovation. Based on ETT efforts in continuous manufacturing, CDER’s Office of Pharmaceutical Quality (OPQ) published a draft guidance, “Quality Considerations for Continuous Manufacturing” of solid oral dosage forms in early 2019.
Under this program, CDER has approved five drug applications utilizing continuous manufacturing for FDF manufacturing, and the first application utilizing 3-D printing technologies. Currently, these drugs are being made in the US, and one drug is being made both in the US and the UK.
“The adoption of advanced manufacturing technologies may pose a challenge to the current regulatory framework because most regulations were developed based on traditional batch manufacturing methods under a unified pharmaceutical quality system,” explained Woodcock in her testimony. “As a result, FDA has launched an effort to identify and implement needed changes in the regulatory structure. For example, new policy and regulatory topics related to emerging technologies include the management of data-rich environments, the evolving concepts of process validation for advanced manufacturing systems, and the regulatory oversight of post-approval changes for such systems.”
Other FDA initiatives include a collaboration with CDER and the Biomedical Advanced Research and Development Authority, whch are working on a strategy and new regulatory framework to develop and implement miniature, mobile manufacturing platforms (i.e., Pharmacy on Demand) for manufacture of essential drugs near or at the point of care. Woodcock also pointed to the FDA’s participation in the work of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) to develop the ICH guideline, Q13, on continuous manufacturing of drug substances and drug products for both small-molecule and biological products to help achieve global regulatory harmonization.




