2025: The Bio/Pharma Industry’s Year in Review

Without question, US-imposed tariffs and ensuing trade deals and the impact on the bio/pharma industry was the number one news story in 2025, going hand-in-hand with US manufacturing investments and drug pricing reforms. What else made the news?
By Patricia Van Arnum, Editorial Director, DCAT, pvanarnum@dcat.org
Tariffs: the story of the year
Tariff/trade policy in the US has been the story of the year as the US government moved forward with plans to impose reciprocal tariffs or country-by-country tariffs as well as laying the groundwork for possible pharmaceutical industry-specific tariffs and the tariff situation continues to loom large.
The Trump Administration first laid out a plan for imposing reciprocal tariffs in February (February 2025) to counter non-reciprocal trading arrangements with US trading partners and to improve US competitiveness, including in manufacturing. Those reciprocal tariffs were scheduled to go into effect on April 9, 2025, but the Administration placed a 90-day pause (until July 9, 2025) on their implementation to enable countries to negotiate these tariffs with the US government. The US government then extended the deadline again, to August 7, 2025, except for certain countries, which were sent letters from the White House that specified their tariffs, as the US government continued to negotiate individual deals with its trading partners. Since that time, the US has struck deals/framework agreements with key trading partners of particular relevance to the pharmaceutical industry, including the European Union, the UK, Switzerland, Japan, China, and India.
Att the top of the tariff battle is a pending decision of the US Supreme Court on the legality of the Administration to impose such tariffs under executive action. The US Supreme Court heard oral arguments in November (November 2025) in two cases to decide whether the current Administration has the legal authority to impose tariffs under the International Emergency Economic Powers Act (IEEPA), which was used to impose reciprocal tariffs on a country-by-country basis. Hearing oral arguments is only the first step in the Supreme Court process as it reviews and considers these cases. Until a decision is reached, the reciprocal tariffs remain in place. A ruling is expected in the coming months.
Pharmaceutical industry tariffs and drug pricing
During this year, the prospect of pharmaceutical industry specific tariffs have loomed large. The legal question over reciprocal tariffs is separate from executive authority to impose industry-specific tariffs. The groundwork for potential industry-specific tariffs was laid earlier this year (April 2025), when the US Department of Commerce initiated an investigation to determine the effects on US national security of imports of pharmaceuticals and pharmaceutical ingredients. The statutory authority for such an investigation comes under Section 232 of the Trade Expansion Act of 1962, as amended, which allows the President to impose import restrictions based on an investigation and affirmative determination by the US Department of Commerce that certain imports threaten to impair US national security. The time frame for this process would require any actions to be taken by the end of this year (2025).
In the meantime, the Trump Administration has struck deals with individual pharmaceutical companies to provide a three-year exemption for Section 252 tariffs in return for commitments for increasing capital investment in the US and agreeing to reduce drug prices for certain products as outlined in individual company agreements to be in line with the Administration’s policy interest for most-favored-nation drug pricing. The premise behind most-favored-nation drug pricing is that the US pays higher prescription drug costs comparative to other developed countries and therefore assumes a larger share of the costs of drugs and that measures should be taken to reduce the differential in the prices of prescription drugs in the US compared to other developed countries, where prescription drug prices are lower. The individual company agreements with the Administration thus far have included deals with Pfizer, AstraZeneca, Eli Lilly and Company, and Novo Nordisk.
These companies were among 19 companies sent letters in July (July 2025) by the Trump Administration to outline the steps they must take to be in alignment with most-favored-nation drug pricing following the issuance of an Executive Order in May (May 2025)) in which the Administration was seeking voluntary compliance with most favored drug pricing. Letters were also sent to AbbVie, Amgen, Boehringer Ingelheim, Bristol Myers Squibb, EMD Serono, Genentech, Gilead Sciences, GSK, Johnson & Johnson, Merck Co., Novartis, , Regeneron, and Sanofi.
US-based bio/pharmaceutical manufacturing
Part of the policy rationale for imposing tariffs is to increase US competitiveness in manufacturing by creating the financial incentive for companies to base operations in the US to avoid tariff burden. This year has seen a slate of announced expansions in the US—greenfield and expansions of existing facilities for manufacturing, research and development, and other capital projects—by the large bio/pharmaceutical companies. These multi-year investments represent new projects as well as existing projects with announcements for future and recent expansions and total investment amounts made by AbbVie ($10 billion), Amgen ($3 billion), AstraZeneca ($50 billion), Biogen ($2 billion), Eli Lilly and Company ($50 billion), Gilead Sciences ($32 billion), GSK ($30 billion), Johnson & Johnson ($55 billion), Merck & Co. ($70 billion), Novartis ($23 billion), Novo Nordisk ($4.1 billion), Pfizer ($70 billion), Regeneron Pharmaceuticals ($2 billion), Roche ($50 billion) and Sanofi ($20 billion).
In addition, the Trump Administration took several steps to facilitate the onshoring of pharma manufacturing into the US. In August (August 2025), the US Food and Drug Administration (FDA) announced the launch of FDA PreCheck, a new program designed to strengthen the domestic pharmaceutical supply chain by increasing regulatory predictability and facilitating the construction of manufacturing sites in the US. FDA PreCheck was developed in response to an Executive Order issued by President Donald Trump in May (May 2025) to streamline FDA review of pharmaceutical manufacturing facilities in the US, ehenhance the inspection process of foreign drug-manufacturing facilities, and streamline permitting of manufacturing facilities by the US Environmental Protection Agency. Also, earlier this year (2025), the President Trump issued an Executive Order to increase the national stockpile for active pharmaceutical ingredients (APIs) of essential medicines. This follows earlier actions under the first Trump Administration. In 2020, the President issued an Executive Order to increase the domestic procurement of essential medicines, medical countermeasures, and critical inputs and to identify supply chain vulnerabilities.
The EU: The Critical Medicines Act
Just as the US is seeking to increase domestic manufacturing through public policy so is the European Union (EU) by proposing measures to reduce shortages of essential/critical medicines by improving security of supply and availability in the EU. In March (March 2025), the European Commission sought to address these issues by publishing a proposal for the Critical Medicines Act, a new EU regulation that aims to improve the availability and security of supply of essential medicines within the EU, including antibiotics, insulin, and painkillers. The proposal seeks to make Europe’s healthcare system more resilient and self-sufficient to better withstand global health challenges that threaten access to essential medicines, such as pandemics, natural disasters and supply chain disruptions. The new rules would complement the proposals to revise the EU’s pharmaceutical legislation, which also contain provisions on managing shortages and the supply of medicinal products.
As part of the legislative process to enact the Critical Medicines Act, this week (December 2, 2025), the Council of the European Union agreed to its position on the Critical Medicines Act, which provide new rules to incentivize supply chain diversification, facilitate collaborative procurement models, and create incentives to boost pharmaceutical manufacturing in EU countries. Once the European Parliament has established its position, the two institutions will enter into negotiations to agree on the final text.
Specifically, the Critical Medicines Act aims to improve the availability, security, and resilience of the supply chain for essential medicines by: diversifying the supply chain to reduce dependence on a limited number of suppliers; providing incentives for collaborative procurement initiatives to help EU member states purchase medicines more effectively and reduce costs; and boosting EU pharmaceutical manufacturing to ensure that critical medicines are produced within the EU by reducing reliance on third countries (i.e., non-EU countries).
The main changes to the Critical Medicines Act introduced by the Council relate to the procurement of critical medicines and their active ingredients. In its position, the Council has:
- Introduced a requirement for the European Commission to issue guidance, including to help EU member states determine whether a critical medicine or active ingredient has been produced in the EU;
- Facilitated the exchange of information on contingency stocks of medicinal products to improve transparency;
- Streamlined the provisions on collaborative procurement and reduced (from nine to six) the minimum number of EU member states needed to submit a joint request to the European Commission to make it easier for countries to work together in securing essential medicines;
- Added an obligation to use criteria relating to resilience in the public procurement of critical medicines to take precedence over price when purchasing critical medicines to ensures procurement decisions prioritize security of supply over costs; and
- Improved the legal clarity and coherence of the regulation by aligning its terminology with that of the public procurement directive.
Change at FDA and product innovation
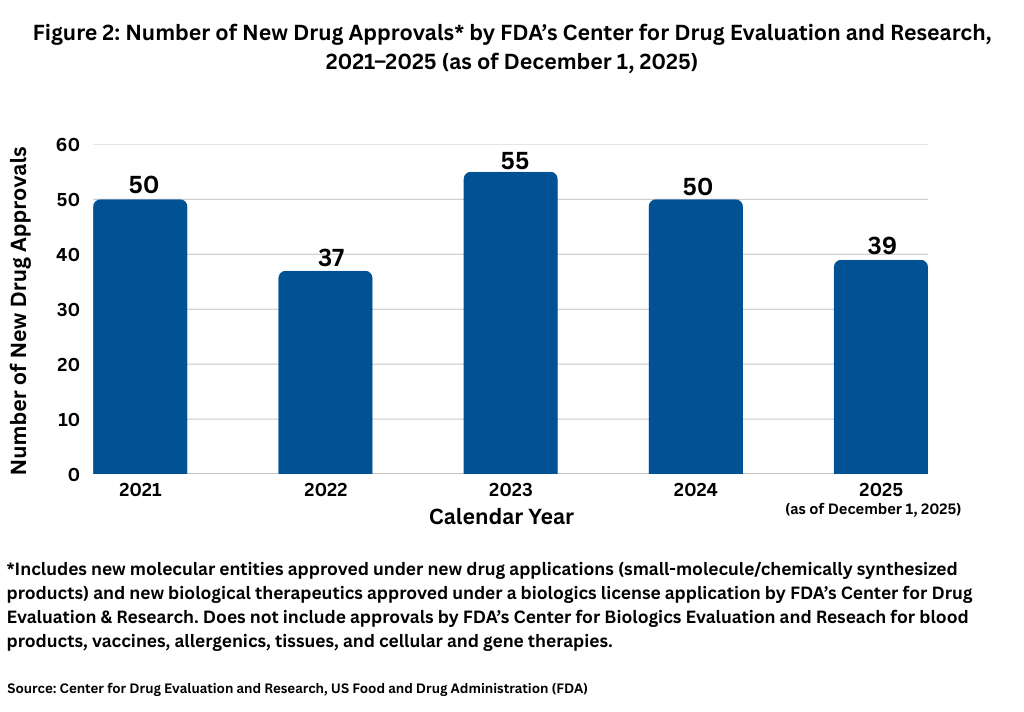
The number of new drug approvals by the US Food and Drug Administration (FDA) is an important barometer of the level of product innovation in the bio/pharma industry, but this year has presented some challenges on the regulatory front. First, staffing and budgetary cuts at the agency, changes in FDA leadership, and a seven-week shutdown of the US government in which reviews of new drug filings were limited have made this year a different environment.
The funding impasse began with the change in the fiscal year on October 1, 2025, after Congress failed to pass legislation to fund the US government. That impasse ended when Congress passed and the President signed into law on November 12, 2025, legislation that funds the US government. With respect to new drug reviews, during the 43-day government shutdown, FDA continued to operate with activities that were funded through carryover user fees and other unlapsed funding. User fee funds specifically support the review and marketing authorization of new medical products. During the funding lapse, FDA was not able to accept new drug applications (NDAs) and biologics license applications (BLAs) that required payment of a user fee although reviews of applications for which a user fee had already been submitted prior to the shutdown could continue. To what extent that this pause will have on the overall number of new drug approvals for 2025 has yet to be seen.
For now, through December 1, 2025, FDA’s Center for Drug Evaluation and Research (CDER) has approved 39 new molecular entities approved under NDAs (small-molecule/chemically synthesized products) and new biological therapeutics, such as recombinant proteins and monoclonal antibodies, approved under a BLA. Other biologic-based products, including blood products, vaccines, allergenics, tissues, and cellular and gene therapies, are reviewed and approved by a separate center within FDA, the Center for Biologics Evaluation and Research (CBER), and are not part of this analysis. Although new drug approvals do not follow a strictly chronological path, with less than a month left in 2025, the level of new drug approvals is off the pace from recent years. Figure 1 shows full-year drug approvals from 2021 to the present, and with the exception of 2022, there have been 50 or more new drug approvals in a given year.
Mergers and acquisitions: some key deals in 2025
Although there were no mega deals in the bio/pharmaceutical in terms of the prescription drug market, there were some key mergers and acquisitions (M&A) as the top companies built their pipelines and portfolios through bolt-on acquisitions.
Top on the list thus far in 2025 is Johnson & Johnson’s $14.6-billion acquisition of Intra-Cellular Therapies, a Bedminster, New Jersey-based bio/pharmaceutical company developing therapeutics for central nervous system disorders. A key asset in the deal is Intra-Cellular Therapies’ Caplyta (lumateperone), a once-daily oral therapy approved to treat adults with schizophrenia, as well as depressive episodes associated with bipolar I or II disorder (bipolar depression), as a monotherapy and adjunctive therapy with lithium or valproate. The drug is also being reviewed as an adjunctive treatment for major depressive disorder with a supplemental new drug application submitted to the US Food and Drug Administration for this indication. The acquisition also included ITI-1284, a Phase II compound being studied in generalized anxiety disorder and Alzheimer’s disease-related psychosis and agitation.
In October (October 2025), Novartis agreed to acquire Avidity Biosciences, an RNA-focused bio/pharmaceutical company, for $12 billion to boost Novartis late-stage neuroscience pipeline. Avidity is developing RNA therapeutics, antibody oligonucleotide conjugates (AOCs), for treating rare genetic neuromuscular diseases. The pending acquisition will provide Novartis with three late-stage candidates: (1) delpacibart etedesiran for treating myotonic dystrophy Type 1, a rare progressive neuromuscular disorder; (2) delpacibart braxlosiran for treating facioscapulohumeral muscular dystrophy,a rare hereditary disorder causing loss of muscle function and progressive disability; and (3) delpacibart zotadirsen for treating Duchenne muscular dystrophy, an early-onset disease marked by progressive muscle damage and reduced life expectancy. The deal does not include Avidity’s early-stage programs in precision cardiology, which is being spun off separately by Avidity. The acquisition is expected to close in the first half of 2026, subject to customary closing conditions, regulatory approvals, and shareholder approvals.
In October (October 2025), Merck & Co. completed its acquisition of Verona Pharma, a bio/pharmaceutical company focused on respiratory diseases, for $10 billion. Through this acquisition, Merck adds Ohtuvayre (ensifentrine), a selective dual inhibitor of phosphodiesterase 3 and 4 (PDE3 and PDE4), to its cardio-pulmonary pipeline and portfolio. The US Food and Drug Administration approved Ohtuvayre in June 2024 for the maintenance treatment of chronic obstructive pulmonary disease in adult patients. Ohtuvayre combines bronchodilator and non-steroidal anti-inflammatory effects. Ohtuvayre is also being evaluated in clinical trials for the treatment of non-cystic fibrosis bronchiectasis.
Last month (November 2025), Merck agreed to acquire Cidara Therapeutics, a San Diego, California-based clinical-stage bio/pharmaceutical company developing drug-Fc conjugate therapeutics, for $9.2 billion. Cidara is using its proprietary Cloudbreak platform to develop drug-Fc conjugates (DFCs) that couple targeted small molecules and peptides to a human antibody fragment (Fc). These single molecule cocktails can be designed to inhibit specific disease targets and to simultaneously engage the immune system, according to information from Cidara. Cidara’s lead candidate, CD388, in Phase III development, consists of a small-molecule neuraminidase inhibitor stably conjugated to a proprietary Fc fragment of a human antibody designed to prevent influenza A and B. The deal is expected to close in the first quarter of 2026.
In July (July 2025), Sanofi completed its acquisition of Blueprint Medicines, a Cambridge, Massachusetts-based bio/pharmaceutical company specializing in rare diseases, in a $9.5-billion deal ($9.1 billion upfront and up to $400 million in milestone payments). Blueprint’s lead commercial product is Ayvakit/Ayvakyt (avapritinib), a rare immunology disease medicine approved in the US and the European Union. The drug had net revenues of $479 million in 2024 and nearly $150 million in the first quarter of 2025. In its first-quarter 2025 earnings results, Blueprint projected full-year 2025 global net product revenues of Ayvakit/Ayvakyt of $700 million to $720 million with potential global net product revenues of $2 billion by 2030. Blueprint’s key pipeline assets include elenestinib for treating systemic mastocytosis, as well as BLU-808, an oral wild-type KIT inhibitor that has the potential to treat a broad range of diseases in immunology, according to the companies.
The larger deals in the bio/pharmaceutical industry in 2025 occurred outside the innovator prescription drug market in the over-the-counter (OTC) and consumer products space. Last month (November 2025), Kimberly-Clark Corporation, a consumer products company, agreed to acquire Kenvue, the former consumer products and OTC healthcare business of Johnson & Johnson (J&J) and now an independent company, for $48.7 billion. The acquisition of Kenvue comes approximately two years after J&J completed its move to demerge its consumer products and OTC business into a separate publicly traded company. J&J initially owned 89.6% of Kenvue following Kenvue’s initial public offering in 2023 but divested its stake through a series of transactions, which were completed in 2024. The transaction is expected to close in the second half of 2026, subject to the receipt of Kenvue and Kimberly-Clark shareholder approvals, regulatory approvals, and satisfaction of other customary closing conditions.
In May (May 2025), Sanofi completed the sale of a 50% controlling stake in its consumer healthcare business, Opella, to CD&R, an investment firm, for EUR 10 billion ($11.4 billion). Following closing, CD&R now has a 50.0% stake in Opella, Sanofi a 48.2% stake, and Bpifrance, a French public sector investment bank, a 1.8% stake.
In the generics/specialty pharma space, Mallinckrodt and Endo, two specialty pharma and generics companies, merged in a $6.7-billion deal, forming a new combined company, Keenova Therapeutics, which consists of the companies’ specialty/brands businesses. After the deal closed, the companies spun off Mallinckrodt’s and Endo’s generics businesses and Endo’s sterile injectables business into a standalone company, Par Health.
Leadership changes
Among key leadership changes, GSK will be headed by a new CEO beginning in 2026, Luke Miels, currently Chief Commercial Officer at GSK, who was appointed CEO Designate, succeeding Emma Walmsley, who will step down as CEO, effective at the end of 2025, with Miels assuming full responsibilities as CEO and joining the Board on January 1, 2026.
Miels joined GSK in 2017, and as Chief Commercial Officer, he has global responsibility for medicines and vaccines. Per the company, he has been instrumental in building GSK’s specialty medicines portfolio, notably in oncology and respiratory. He is an experienced global biopharma leader, having worked at senior levels in the US, Europe, and Asia, and at AstraZeneca, Roche and Sanofi-Aventis (now Sanofi), prior to joining GSK.
Walmsley joined the GSK Board as CEO Designate on January 1, 2017 and became CEO on April 1, 2017. She has been a member of the GSK Leadership Team since 2011. Before being appointed as GSK’s CEO, she was the CEO of GSK Consumer Healthcare, a joint venture between GSK and Novartis, from its creation in March 2015. She joined GSK in 2010 from L’Oreal, having worked there for 17 years in a variety of roles in Paris, London, New York, and Shanghai.
Novo Nordisk named a new President and CEO, Mike Doustdar, who took over the helm at the company in early August (August 2025), succeeding then CEO and President Lars Fruergaard Jørgensen. The company had announced in May (May 2025) that Jørgensen would be stepping down, then citing a need for a change in executive leadership amid challenging conditions. The company faced increased competition from its blockbuster drugs, Ozempic/Wegovy (semaglutide), glucagon-like peptide 1 (GLP-1) agonists, respectively for treating Type 2 diabetes and obesity, potentially less than optimal results for its next-generation treatments, and a sharp decline earlier this year (2025) in the company’s stock price, according to analysts.
In addition, Merck KGaA, Darmstadt, Germany, appointed Kai Beckmann, current CEO of the company’s Electronics Business, as future Chair of the Executive Board and CEO. He will take over this position on May 1, 2026, from Belén Garijo, current CEO and Chair of Merck KGaA, who will continue to lead the company until the end of her tenure.
The battle in the obesity drug market
The battle in the obesity drug market was a key story in 2025 marked by bidding wars in mergers and acquisitions, restructuring and leadership changes at Novo Nordisk, and a race to have the first oral GLP-1 agonist for treating obesity.
The leaders now in the obesity drug market are Novo Nordisk and Eli Lilly and Company, who battle one-to-one in both the diabetes and obesity market with Novo’s Ozempic/Wegovy (semaglutide), a GLP-1 agonist, and Lilly’s Mounjaro/Zepbound (tirzepatide), a dual-activating GIP (glucose-dependent insulinotropic polypeptide) and GLP-1 medication, respectively for treating Type 2 diabetes and obesity. For both Novo Nordisk’s Ozempic/Wegovy and Lilly’s Mounjaro/Zepbound, the obesity indication is a key driver in realizing blockbuster status for these drugs, which are administered as injectables. Both companies also are moving forward with advancing oral obesity drugs.
Novo Nordisk is advancing an oral version of its obesity drug, Wegovy. Novo Nordisk already has an oral version of semaglutide, the active ingredient in Ozempic and Wegovy, on the market in Rybelsus, which was first approved in 2019, but that is indicated for treating Type 2 diabetes, not obesity. In May (May 2025), Novo announced that the US Food and Drug Administration (FDA) accepted its new drug application (NDA) submission for an investigational once-daily oral formulation of Wegovy (semaglutide) for chronic weight management in adults living with obesity or overweight with one or more comorbid conditions and to reduce the risk of major adverse cardiovascular events in adults with overweight or obesity and established cardiovascular disease. If approved, Wegovy would become the first oral formulation of a GLP-1 indicated for chronic weight management. The FDA action date to decide on the Wegovy oral formulation NDA is in the fourth quarter of 2025.
Lilly is advancing orforglipron, a once-daily oral small-molecule (non-peptide) oral GLP-1 receptor agonist, for treating Type 2 diabetes and obesity. Orforglipron was discovered by Chugai Pharmaceutical and licensed by Lilly in 2018. Chugai and Lilly published the preclinical pharmacology data of this molecule together, and Lilly is running Phase III studies on orforglipron for the treatment of Type 2 diabetes and for weight management in adults with obesity or overweight with at least one weight-related medical problem. It is also being studied as a potential treatment for obstructive sleep apnea and hypertension in adults with obesity. The company expects to submit to global regulatory agencies orforglipron in obesity by the end of 2025.
The lure of the obesity drug market was the impetus in a bidding war between Pfizer and Novo Nordisk for acquiring Metsera, a New York-based clinical-stage bio/pharmaceutical company focused on obesity and cardiometabolic diseases, with Pfizer emerging victorious with a successful offer, worth up to $10 billion, in a deal that closed last month (November 2025). Pfizer’s move to acquire Metsera followed recent setbacks in its in-house drug candidates for obesity. Pfizer discontinued development of two GLP-1 RAs: lotiglipron in 2023 and danuglipron in April of 2025.
For its part, Novo Nordisk’s now unsuccessful move to acquire Metsera leaves the company with a growth strategy still focused on diabetes and obesity as it navigates increased competition from its blockbuster drugs, Ozempic/Wegovy (semaglutide), respectively for treating Type 2 diabetes and obesity, and as it seeks to gain traction for its next-generation products. In September (September 2025), Novo Nordisk announced a company-wide restructuring to reduce its global workforce by approximately 9,000 positions, a reduction of 11.5%. The restructuring was the first major move by Novo Nordisk’s new President and CEO Mike Doustdar, who took over the helm at the company in early August (August 2025), succeeding then CEO and President Lars Fruergaard Jørgensen, who had announced in May (May 2025) that he would be stepping down.




