Brexit: Are Pharma Companies Prepared?

A recent European Medicines Agency (EMA) survey shows that marketing authorization holders for more than half (58%) of the 694 centrally authorized products in the UK are on track with their regulatory planning to ensure that their marketing authorization remains valid once the UK leaves the European Union.
However, for 108 (88 human products and 20 veterinary products), or 16%, of these medicines with manufacturing sites located in the UK only, there are serious concerns that the necessary actions will not be carried out in time. DCAT Value Chain Insights examines pharmaceutical companies’ preparedness for Brexit.
Brexit and the pharmaceutical industry
The question on how the UK should exit the EU (i.e., Brexit) and the resulting trade and custom policies that would be part of such a plan is the subject of intense debate in the UK between those that wanted a clean break with the EU and those who favor a so-called “softer” exit that would keep some economic ties with the EU. Last week (on July 12, 2018), the UK government issued a white paper, The Future Relationship between the UK and the European Union (EU), that detailed a new trading relationship under which the UK is emphasizing that the UK and the EU should ensure what it calls “continued frictionless access at the border to each other’s markets for goods.”
To deliver this goal, the UK government is proposing the establishment of a free trade area for goods. The proposal would avoid the need for customs and regulatory checks at the border and mean that businesses would not need to complete customs declarations and would enable products to only undergo one set of approvals and authorizations in either market before being sold in both. These close arrangements on goods would sit alongside new ones for services and digital to provide what the UK government calls “the freedom to chart its own path in the areas that matter most for its economy,” said the report.
“The [UK] Government wants to minimize new barriers to trade between the UK and the EU and hopes that both sides will work together to reduce them further over time—but acknowledges that there will be more barriers to the UK’s access to the EU market than is the case today,” said the white paper. “…[A] relationship this deep will need to be supported by provisions giving both sides confidence that the trade that it facilitates will be both open and fair. So the [UK] Government is proposing reciprocal commitments that would ensure UK businesses could carry on competing fairly in EU markets, and EU businesses operating in the UK could do the same.”
Amidst parliamentary battles concerning the details of the UK’s proposal for a trade policy under Brexit, the UK Parliament voted in favor of an amendment to a trade bill that will make it a negotiating objective for the UK government to seek the UK’s participation in the European medicines regulatory network, a move that is supported by the UK pharmaceutical industry.
In a joint statement on behalf of the pharmaceutical industry in the UK, Mike Thompson, Chief Executive of the Association of the British Pharmaceutical Industry, which represents innovator, research-based companies in the UK, and Steve Bates, Chief Executive of the BioIndustry Association, which represents the biotechnology industry in the UK, issued a joint statement to support the recent action by the UK Parliament to prioritize the UK’s position with the framework of European Medicines Agency (EMA), the pharmaceutical regulatory body of the EU, post-Brexit.
“Today, Parliament has sent a clear message that patients and public health should be a top priority for the Government in these negotiations,” they said in the July 17, 2018 joint statement. “Every month, 37 million packs of medicine arrive in the UK from the EU and 45 million move the other way. Therefore, it is essential that the UK continues to participate in the EMA after Brexit as set out in the Brexit White Paper and in the Prime Minister’s Mansion House speech.”
Brexit preparedness: marketing authorizations
As the debate for a trade and customs policy post Brexit continues, the EMA released the results of a study that examined how prepared the pharmaceutical industry is for Brexit. If Brexit proceeds as scheduled, the UK will exit the EU on March 29, 2019 (11 pm UK time), with the first full day of post-Brexit on March 30, 2017. Since May 2017, the European Commission and the EMA have informed companies and raised their awareness of the need to put the necessary measures in motion to keep their marketing authorizations valid, according to information from the EMA. Information notices on legal issues and guidance on practical and simplified requirements for companies have been published and regularly updated according to the EMA.
For marketing authorization holders of centrally authorized products, this may imply changes to the marketing authorization. Currently, under the centralized authorization procedure, pharmaceutical companies submit a single marketing-authorization application to the EMA, which if approved, allows the marketing-authorization holder to market the approved medicine and make it available to patients and healthcare professionals throughout the EU on the basis of a single marketing authorization. The EMA’s Committee for Medicinal Products for Human Use or Committee for Medicinal Products for Veterinary Use carry out a scientific assessment of the application and give a recommendation on whether the medicine should be marketed or not. Once granted by the European Commission, the centralized marketing authorization is valid in all EU member states as well as in the European Economic Area (EEA) countries of Iceland, Liechtenstein, and Norway. For marketing authorization holders of centrally authorized products post Brexit, this may imply changes to the marketing authorization itself, including, for example, a transfer of the marketing authorization to a legal entity established in the EEA (which consists of the EU and Iceland, Liechtenstein, and Norway) or a change of the qualified person for pharmacovigilance (QPPV) or pharmacovigilance system master file (PSMF) to a location in the EEA as well as adaptations to their logistics, manufacturing sites, supply chains and contracts.
The EMA launched a survey in January 2018 to identify centrally authorized products that are potentially at risk of supply shortages and to obtain information on the timelines for submission of the necessary regulatory changes to ensure those products remain in supply post Brexit. The survey was sent to marketing authorization holders of the 694 centrally authorized products (661 human and 33 veterinary products) who are located in the UK or who have quality control, batch release and/or import or manufacturing sites, or a QPPV or PSMF in the UK. According to EU law, the marketing authorization holder, the QPPV, the PSMF and certain manufacturing sites need to be based in the EEA for a company to be able to market a medicine in the EU.
The survey found that marketing authorization holders for more than half (58%) of the 694 centrally authorized products with an important step in their regulatory processes in the UK are on track with their regulatory planning to ensure that their marketing authorization remains valid once the UK leaves the EU. However, for 108 (88 human products and 20 veterinary products), or 16%, of these medicines with manufacturing sites located in the UK only, the EMA reported that there are “serious concerns” that the necessary actions will not be carried out in time.
Specifically, EMA databases indicate that 400 centrally authorized products have their marketing authorization holders registered as being established in the UK. Such marketing authorizations will, therefore, need to be transferred and implemented before March 30, 2019, the first full date of post Brexit, to a holder established in the EU/EEA. Out of those targeted 400 products, the marketing authorization holders of 373 of these products have informed the EMA of their intention to submit such transfer application of marketing authorization within the needed deadlines (see Figure 1 ). At the time of completion of this survey, marketing authorization holders of 335 centrally authorized medicinal products (both human and veterinary) have indicated that their QPPVs are based in the UK. For the PSMF (for human medicines only), 376 are based in the UK. Based on responses received, 282 (84%) and 152 (40%) of QPPVs and PSMFs, respectively, were confirmed as on track for the requisite changes to be made by the deadline of March 30, 2019 (see Figure 1).
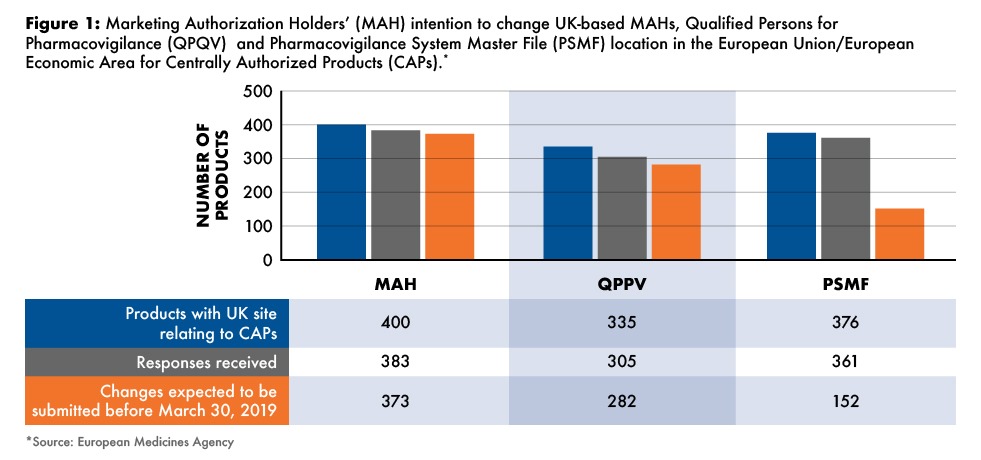
Overall, the EMA has said that the industry has been active in planning such transfers in advance of the UK withdrawal from the EU/EEA for human medicinal products. According to the responses received from the survey, the large majority of companies (94%) plan to submit transfer applications in due time. For 27 centrally authorized medicinal products (6%), no responses were received. The EMA said it will contact these marketing authorization holders to obtain feedback on their status of readiness.
In terms of intended submission dates, the majority of transfers for human medicinal products are expected to have taken place by the second quarter of 2018, but such transfers will continue through the fourth quarter of 2018 and the first quarter of 2019 (see Figure 2) when the EMA will be relocating to Amsterdam.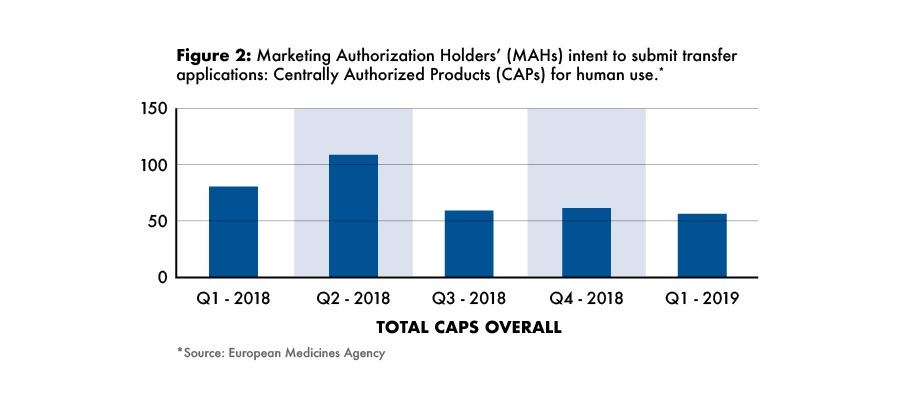
Brexit preparedness: batch-release and site-location changes
To control the quality of the medicinal products circulating within the EU, the Qualified Person of the manufacturing and importation authorization holder is responsible for certifying that each batch of medicinal product intended to be placed on the EEA market was manufactured in accordance with EU good manufacturing practice (GMP) requirements and the marketing authorization. The batch-release site has to be located in the EU/EEA. By the time of the UK withdrawal date from the EU, the marketing authorization holder will, therefore, need to transfer any current UK-based site of batch release to a location established in the EU.
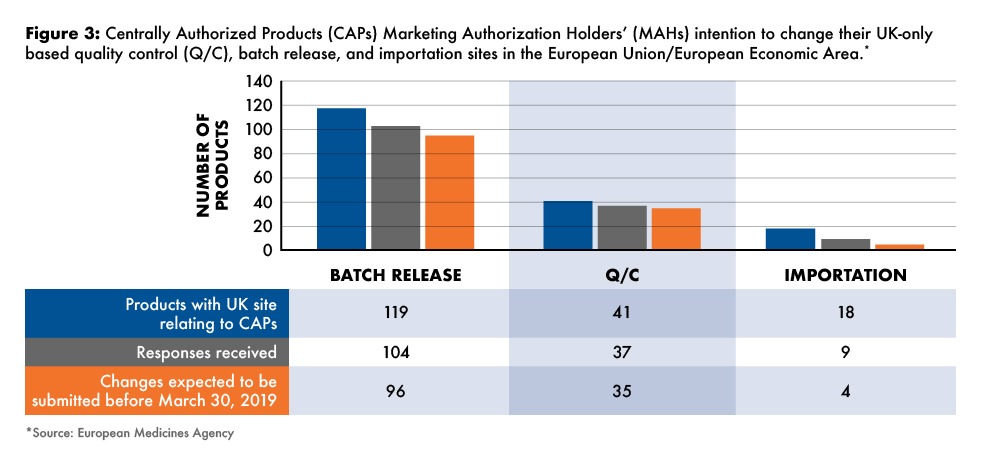
From the responses received to the EMA survey, the EMA was informed that the large majority of the batch-release sites currently located in the UK would be changed to a site in the EU/EEA in due time, representing 96 of 119, or 81%, of the batch sites. Twenty-three, or 19%, of the variations needed to change the batch-release sites currently located in the UK to another EU/EEA member site might not be in time for the changes to be in place prior to March 30, 2019 (see Figure 3) . This includes 15 products for which no response was received from the EMA survey.
According to EU requirements, the marketing authorization holder will need to specify a site of batch control in the EU/EEA where each production batch can undergo, upon importation, a full qualitative analysis, a quantitative analysis of at least all the active substances, and all the other tests or checks necessary to ensure the quality of medicinal products in accordance with the requirements of the marketing authorization. For centrally authorized medicinal products, the marketing authorization will need to change the location of any current UK-based site of batch control to a location established in the EU/EEA and submit the corresponding variation. Further to the analysis of the EMA survey responses, 41 variations for quality-control changes need to be submitted. For 35 (85%) of these medicines, marketing authorization holders are planning submission in due time and six marketing authorization holders, or 15%, have either not responded or indicated submission at a later time point (see Figure 3).
Brexit preparedness: importation-site locations
As of the date of the withdrawal of the UK from the EU, medicinal products manufactured in the UK will be considered imported medicinal products. Manufacturing authorization holders are obliged to use, as starting materials, only active substances that have been manufactured in accordance with the detailed guidelines on GMP for starting materials. Active substances for medicinal products for human use shall only be imported into the EU/EEA if the active substances are accompanied by a written confirmation that GMP standards equivalent to those in the EU/EEA where carried out in the plant in which the imported product was made and that the import of of medicinal products is carried out in accordance with EU requirements. Marketing authorization holders, therefore, will need to specify an authorized importer established in the EU/EEA and submit the corresponding variation. The EMA survey analysis for centrally authorized products showed that of the 18 importation sites identified as needing to be changed from the UK to a new site in the EU/EEA, responses were not received for half of these. Of the nine responses, only four (22%) indicated that they will be submitting before the March 2019 deadline (see Figure 3 ).
In addition, there are 139 and 201 centrally authorized human and veterinary medicinal products that have respectively batch-release and quality-control sites located in the EU/EEA and also in the UK. For 106 of these products, changes to their batch-release or quality-control testing sites might not be submitted before March 29, 2019, according to the EMA. The majority of the changes related to the above-mentioned batch release, quality control and importation site changes are expected to be submitted as minor variations, except when related to biological products, according to the EMA.
Looking ahead
The EMA said it is liaising directly with the marketing authorization holders who either did not reply to the survey or have indicated in the survey that they do not plan to submit the changes required by March 30, 2019 and have manufacturing sites in the UK only, as this could potentially lead to supply disruptions. The EMA said it has analyzed feedback from the survey and is now looking in detail at those medicines where there are risks of supply shortages and will assess how critical these are. EMA said its role is to ensure that it has a complete overview of the potential risks and to work together with the relevant marketing authorization holders to address these risks as early as possible and discuss relevant mitigation measures. The EMA said it will also regularly monitor the submission of changes to marketing authorizations for all 694 products to check if the relevant variations/notifications are being submitted.
The EMA said that a high level of uncertainty also still exists as to where the new sites for PSMF, QPPV and batch release will be located post-Brexit, and consequently further information will be needed for the EMA and EU network inspectorates to plan their workload and resources. The EMA said it will follow up directly with the marketing authorization holders that have batch-release, quality-control and/or importation sites located in the UK only and that have indicated in the EMA survey that they do not plan to submit the changes required before March 30, 2019 as this could potentially lead to supply disruptions. In addition, the EMA said it will monitor and track the submissions of required changes for the affected centrally authorized medicines, and workload analysis will be used to ensure adequate resource planning within the EMA and the network, where relevant. The EMA is “strongly advising” pharmaceutical companies to submit the necessary changes for the continued maintenance of their marketing authorizations to the EMA as early as possible and before the end of fourth quarter of 2018 to ensure processing in due time.